分子对接简要介绍

分子对接简介
分子对接(molecular docking)是通过研究小分子配体与受体生物大分子相互作用,预测其结合模式和亲和力进而实现基于结构的药物设计的一种重要的方法。其本质是两个或多个分子之间的识别过程,其过程涉及分子之间的空间匹配和能量匹配。
分子对接的基本原理
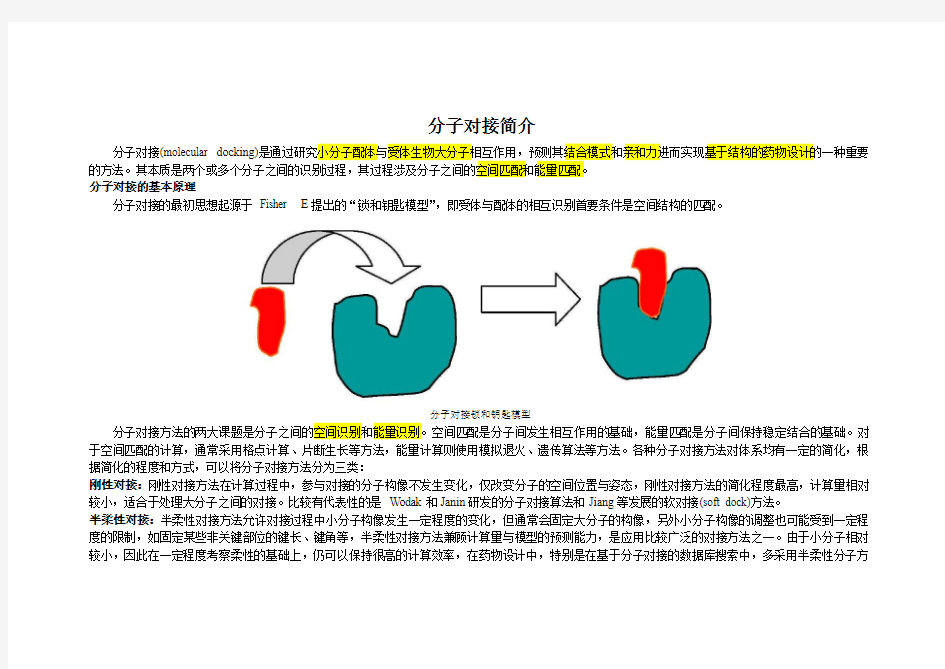
分子对接的最初思想起源于Fisher E提出的“锁和钥匙模型”,即受体与配体的相互识别首要条件是空间结构的匹配。
分子对接锁和钥匙模型
分子对接方法的两大课题是分子之间的空间识别和能量识别。空间匹配是分子间发生相互作用的基础,能量匹配是分子间保持稳定结合的基础。对于空间匹配的计算,通常采用格点计算、片断生长等方法,能量计算则使用模拟退火、遗传算法等方法。各种分子对接方法对体系均有一定的简化,根据简化的程度和方式,可以将分子对接方法分为三类:
刚性对接:刚性对接方法在计算过程中,参与对接的分子构像不发生变化,仅改变分子的空间位置与姿态,刚性对接方法的简化程度最高,计算量相对较小,适合于处理大分子之间的对接。比较有代表性的是Wodak和Janin研发的分子对接算法和Jiang等发展的软对接(soft dock)方法。
半柔性对接:半柔性对接方法允许对接过程中小分子构像发生一定程度的变化,但通常会固定大分子的构像,另外小分子构像的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等,半柔性对接方法兼顾计算量与模型的预测能力,是应用比较广泛的对接方法之一。由于小分子相对较小,因此在一定程度考察柔性的基础上,仍可以保持很高的计算效率,在药物设计中,特别是在基于分子对接的数据库搜索中,多采用半柔性分子方
法。其代表性软件是DOCK和AutoDock。
柔性对接:柔性对接方法在对接过程中允许研究体系的构像发生自由变化,由于变量随着体系的原子数呈几何级数增长,因此柔性对接方法的计算量非常大,消耗计算机时很多,适合精确考察分子间识别情况。其中比较有代表性的方法有Accelrys 公司发展的基于分子力学和分子动力学的分子对接方法及Affinity 软件。
打分函数:
现有打分函数有要有三种:
基于分子力场的打分函数:该方法只考虑只考虑热焓对能量的贡献,不考虑熵的影响,一般情况下,采用标准力场的非键作用能如真空静电和范德华作用能用作打分函数。
基于经验的回归参数的打分函数:用多元回归的方法拟合各种物理参数对结合自由能的贡献。如FlexX 程序中采用这种函数,所采用的方程包括配体旋转键的个数!氢键、离子键、疏水和芳香环的堆积作用,以及亲水作用"这种方法能快速直接地估算结合自由能。
基于知识的打分函数:最初应用于蛋白质结构预测,打分函数用统计力学的方法来自蛋白质-配体的复合物结构,结合自由能用函数为分子间距离的平均能的加和来计算"基于知识的打分函数是一种比较有前途的方法。
各种常用分子对接软件对比
名称优化方法评价函数速度准确度优点缺点使用方式
AutoDock Lamarckian genetic algorithm
Monte Carlo simulated annealing
Traditional genetic algorithm 半经验自由能评价函数一般good accuracy -- -- 免费获取
license
GOLD genetic search algorithm 半经验自由能评价函数快good accuracy -- -- 2个月试
用
FlexX fragment-based 半经验自由能评价函数快-- 比AutoDock 、
GOLD快不适用于
柔性配基
6周的试
用版
ICM global minimum of the energy 半经验自由能评价函数快This program provided the
highest accuracy in ligand
docking against AutoDock,
DOCK, FlexX, and GOLD
in a study involving 37 虚拟筛选能力比DOCK、FlexX 优越
receptors.
DOCK 片断生长分子力场、表面匹配得分、
化学环境匹配得分快低shown to handle
well small
binding
sites, opened
cavities and
small
hydrophobic
ligands
不适用于
柔性配
基、极性
配基
Glide 系统搜寻半经验自由能评价函数快高-- -- --
分子筛知识概述
分子筛知识概述 (一)分子筛的品种型号 分子筛(又称合成沸石)是一种硅铝酸盐多微孔晶体,它是由SiO和AIO四面体组成和框架结构。在分子筛晶格中存在金属阳离子(如Na,K,Ca等),以平衡四面体中多余的负电荷。分子筛的类型按其晶体结构主要分为:A型,X型,Y型等 A型:主要成分是硅铝酸盐,孔径为4A(1A=10-10米),称为4A(又称纳A型)分子筛;用Ca2+交换4A分子筛中的Na+,形成5A的孔径,即为5A(又称钙A型)分子筛;用K+交换4A 分子筛的Na+,形成3A的孔径,即为3A(又称钾A型)分子筛。 X型:硅铝酸盐的晶体结构不同(硅铝比大小不一样),形成孔径为9—10A的分子筛晶体,称为13X(又称钠X型)分子筛;用Ca2+交换13X分子筛中的Na+,形成孔径为9A的分子筛晶体,称为10X(又称钙X型)分子筛 Y型:Y型分子筛具有X型分子筛烃似的晶体结构,但化学组成不同(硅铝比较大)通常用于催化领域。 (二)分子筛的主要特性 1、物理特性: 比热:约0.95KJ/KgXK(0.23Kcal/KgX℃ 导热系数(脱水物):2.09KJ/MXK(0.506Kcal/mX℃ 水吸附热:约3780KJ/Kg(915Kcal/Kg) 2、热稳定性和化学稳定性: 分子筛能承受600—700℃的短暂高温,但再生温度一般在400℃以下。分子筛可在PH值5-10范围的介质中使用;在盐溶液中能交换某些金属阳离子。 3、分子筛的特性 分子筛是一类结晶的硅铝酸盐,由于它具有均一的孔径和极高的比表面积,所以具有许多优异的特点。(1)按分子的大小和形状不同的选择吸附作用,即只吸附那些小于分子筛孔径的分子。(2)对于小的极性分子和不饱和分子,具有选择吸附性能,极性越大,不饱和度越高,其选择吸附性越强。(3)具有强烈的吸水性。哪怕在较高的温度、较大的空速和含水量较低的情况下,仍有相当高的吸水容量。 3.1、基本特性: a)分子筛对水或各种气,液态化合物可逆吸附及脱附。 b)金属阳离子易被交换。
分子动力学模拟方法概述(精)
《装备制造技术》 2007年第 10期 收稿日期 :2007-08-21 作者简介 :申海兰 , 24岁 , 女 , 河北人 , 在读研究生 , 研究方向为微机电系统。 分子动力学模拟方法概述 申海兰 , 赵靖松 (西安电子科技大学机电工程学院 , 陕西西安 710071 摘要 :介绍了分子动力学模拟的基本原理及常用的原子间相互作用势 , 如Lennard-Jones 势 ; 论述了几种常用的有限差分算法 , 如 Verlet 算法 ; 说明了分子动力学模拟的几种系综及感兴趣的宏观统计量的提取。关键词 :分子动力学模拟 ; 原子间相互作用势 ; 有限差分算法 ; 系综中图分类号 :O3 文献标识码 :A 文章编号 :1672-545X(200710-0029-02 从统计物理学中衍生出来的分子动力学模拟方法 (molec- ular dynamics simulation , M DS , 实践证明是一种描述纳米科技 研究对象的有效方法 , 得到越来越广泛的重视。所谓分子动力学模拟 , 是指对于原子核和电子所构成的多体系统 , 用计算机模拟原子核的运动过程 , 从而计算系统的结构和性质 , 其中每一个原子核被视为在全部其他原子核和电子所提供的经验势场作用下按牛顿定律运动 [1]。它被认为是本世纪以来除理论分析和实验观察之外的第三种科学研究手段 , 称之为“计算机实验” 手段 [2], 在物理学、化学、生物学和材料科学等许多领域中得到广泛地应用。
根据模拟对象的不同 , 将它分为平衡态分子动力学模拟 (EM DS (和非平衡态分子动力学模拟 (NEM DS 。其中 , EM DS 是分子动力学模拟的基础 ; NEM DS 适用于非线性响应系统的模拟 [3]。下面主要介绍 EM DS 。 1分子动力学方法的基本原理 计算中根据以下基本假设 [4]: (1 所有粒子的运动都遵循经典牛顿力学规律。 (2 粒子之间的相互作用满足叠加原理。 显然这两条忽略了量子效应和多体作用 , 与真实物理系统存在一定差别 , 仍然属于近似计算。 假设 N 为模拟系统的原子数 , 第 i 个原子的质量为 m i , 位置坐标向量为 r i , 速度为 v i =r ? i , 加速度为 a i =r ?? i , 受到的作用力为 F i , 原子 i 与原子 j 之间距离为 r ij =r i -r j , 原子 j 对原子 i 的作用力为 f ij , 原子 i 和原子 j 相互作用势能为 ! (r ij , 系统总的势能为 U (r 1, r 2, K r N = N i =1! j ≠ i ! " (r ij , 所有的物理量都是随时 间变化的 , 即 A=A (t , 控制方程如下 : m i r ?? i =F i =j ≠ i
分子动力学的模拟过程
分子动力学的模拟过程 分子动力学模拟作为一种应用广泛的模拟计算方法有其自身特定的模拟步骤,程序流程也相对固定。本节主要就分子动力学的模拟步骤和计算程序流程做一些简单介绍。 1. 分子动力学模拟步驟 分子动力学模拟是一种在微观尺度上进行的数值模拟方法。这种方法既可以得到一些使用传统方法,热力学分析法等无法获得的微观信息,又能够将实际实验研究中遇到的不利影响因素回避掉,从而达到实验研宄难以实现的控制条件。 分子动力学模拟的步骤为: (1)选取所要研究的系统并建立适当的模拟模型。 (2)设定模拟区域的边界条件,选取粒子间作用势模型。 (3)设定系统所有粒子的初始位置和初始速度。 (4)计算粒子间的相互作用力和势能,以及各个粒子的位置和速度。 (5)待体系达到平衡,统计获得体系的宏观特性。 分子动力学模拟的主要对象就是将实际物理模型抽象后的物理系统模型。因此,物理建模也是分子动力学模拟的一个重要的环节。而对于分子动力学模拟,主要还是势函数的选取,势函数是分子动力学模拟计算的核心。这是因为分子动力学模拟主要是计算分子间作用力,计算粒子的势能、位置及速度都离不开势函数的作用。系统中粒子初始位置的设定最好与实际模拟模型相符,这样可以使系统尽快达到平衡。另外,粒子的初始速度也最好与实际系统中分子的速度相当,这样可以减少计算机的模拟时间。 要想求解粒子的运动状态就必须把运动方程离散化,离散化的方法有经典Verlet算法、蛙跳算法(Leap-frog)、速度Veriet算法、Gear预估-校正法等。这些算法有其各自的优势,选取时可按照计算要求选择最合适的算法。 统计系统各物理量时,便又涉及到系统是选取了什么系综。只有知道了模拟系统采用的系综才能釆用相对应的统计方法更加准确,有效地进行统计计算,减少信息损失。 2. 分子动力学模拟程序流程 具体到分子动力学模拟程序的具体流程,主要包括: (1)设定和模拟相关的参数。 (2)模拟体系初始化。 (3)计算粒子间的作用力。 (4)求解运动方程。 (5)循环计算,待稳定后输出结果。 分子动力学模拟程序流程图如2.3所示。
分子筛的一些知识
分子筛的一些知识 沸石分子筛的广泛应用,在于它具有优异的性能。为了深刻了解这些性能,就必须弄清分子筛的结构,而深入研究分子筛的结构与性能,反过来又将进一步促进它的应用和发展。 分子筛是一种晶体硅铝酸盐,因而,可以应用X-射线衍射进行结构分析。通常合成分子筛是10μ以下的粉末,在使用粉末衍射法进行测试时,对于对称性较差的沸石类型,指标化及搜集强度的工作都十分困难,因此,到目前为止,仅确定了四十多种沸石的结构,还有一半左右尚未测定出来。 倘若能够得到较大的佛石单晶,采用X-射线衍射的单晶转动法更为有效。较大的A型分子筛单晶可由种子晶体的再结晶得到。用X-射线衍射的单晶转动法,不仅可在指标化之前,引出晶胞参数,确定骨架结构,而且还可以推定出非骨架原子(或离子)和分子和位置。每一种分子筛都有特征的X-射线粉末衍射图样,因此由衍射图样的比较,可以确定沸石的类型。非晶态度的凝胶不产生衍射,故X-射线分析也可以用来观察分子筛结晶的情况,混和物的衍射图样,由各组分的粉末线迭合而成,并且衍射强度随含量而变化。所以X-射线衍射也用以确定分子筛的纯度。 现在又有一种新的红外光谱法测定分子筛的结构。通过测定已知结构分子筛的红外光谱,找出普带的特征频率与骨架结构基团间的关系,进而测定未知结构的光谱,推导出骨架结构。一般采用频率1300-200厘米-1的红外线。因为这一范围包含着(Si,Al)O4四面体的基本振动,反映出骨架结构的特征。目前,红外光谱已用于测定沸石骨架的结构类型,结构基团、骨架的硅铝组成,热分解过程中结构的变化和脱水、脱羟基过程中阳离子的迁移等。 在分子筛的应用中,表面性质十分重要。借助红外光谱,我们可以更透彻地了解沸石的表面性质以及在各种处理中的变化,如根据吸附分子引起的光谱变化,就可知道沸石表面与吸附分子相互作用,吸附分子的位置以及催化活性和表面性质的关系等。由于红外光谱的高度灵敏性,沸石结构的微小变化都在光谱中得到反映。 其他的物理测试技术如紫外光谱等也可以帮助确定分子筛的结构,但目前主要采用的是X-射线衍射和红外光谱法。 沸石A、沸石X、沸石Y和丝光沸石应用最广,对它们的结构和性能的研究也最为深刻。第一节分子筛结构概述 分子筛是一类具有骨架结构的硅铝酸盐晶体,晶体内的阳离子和水分子在骨架中有很大的移动自由度,可进行阳离子交换和可逆地脱水。 分子筛的化学组成可用以下实验式表示:M2/nO. Al2O3. xSiO2. yH2O M是金属离子,n是M的价数,x是SiO2.的分子数,也是SiO2/Al2O3克分子比,y是水分子数. 上式可以改写成M p/n[(AlO2)p()q] yH2O P是AlO2分子数,q是SiO2分子数,M,n,y同上.由上式可以看出:每个铝原子和硅原子平均加起来都有二个氧原子,若金属原子M的化合价n=1,则M的原子数等于铝原子数,若n=2,则M 的原子数等于铝原子数的一半。各种分子筛的区别,首先是化学组成的不同,如经验式中的M可为Na、K、Li、Ca、Mg等金属离子,也可以是有机胺或复合离子。 化学组成的一个重要区别是硅铝克分子比的不同。例如,沸石A、沸石X、沸石Y和丝光沸石的硅铝比分别为1.5~2、2.1~3.0、3.1~6.0和9~11。 当式中的x数值不同时,分子筛的抗酸性、热稳定性以及催化活性等都不同,一般x的数值越大,而酸性和热稳定性越高。各种分子筛最根本的区别是晶体结构的不同,因而,不同的分子筛具有不同的性质。
高校学生入党积极分子自我介绍范文
高校学生入党积极分子自我介绍范文 我叫xx,19xx年x月x日出生于xx市的一个普通家庭,xxxx年加入共青团组织,并于xxxx年底递交了入党申请书,现在就读于xx大学xx系xx班。我志愿加入中国共产党,成为一名光荣的共产党员。因为中国共产党是中国工人阶级的先锋队,是中国各族人民利益的忠实代表,是中国社会主义事业的领导核心。党的最终目标是实现共产主义的社会制度。作为一名入党积极分子,首先要解决为什么要入党的问题。因为入党动机是激励我入党的主观原因,从根本上决定了作为一名党员应具有的素质和行为,也是个人世界观、人生观的集中反映。其实正确的入党动机的形成并不在一朝一夕,而是在我三年多的学习、思考中渐渐地体会到的。 下面我详细地就我的入党动机问题谈一下我的心理历程。刚递交入党申请书时,是因为看到别人写入党申请,如果自己不写,怕老师,同学说自己不要求进步,于是也随大流写份申请,但是其间的所写却都是从书上,从党员父亲那里抄来、学来的。其实那时的我对党缺乏真正的认识,尽管上过几次党课,但对党的基本知识还是知之甚少,思想上并没有迫切要求入党的愿望。这是一种对党、对个人都不认真、不严肃的政治态度。后来到高中毕业前夕,看到有的同学入了党,在同学中有一定的威望,羡慕不已,于是自己赶紧写了几份思想汇报,企盼有一天自己能获此荣耀,得到精神上的满足。其实这是一种入党动机不纯的表现。入党不是给别人炫耀的,更不是用来满足自己虚荣心的,而是应该踏踏实实为党奉献,全心全意为人民服务,在学习和生活中发挥先锋模范作用。 大一时,在上党的基本知识概论课中学习了党的基本知识,同时有更多的时间和机会接触到身边的党员,政治视野也得到了扩充,看到当前存在党风不正的现象,希望自己能加入到党组织,做一名优秀的党员,重新确定党的形象。这种看法现在想起来是很片面和狭隘的,这种错误想法的产生究于对党的认识还太模糊,不能以因为有些党员存在着这样那样的缺点就否定他们是无产阶级的先锋战士,一直影响对党性的认识,也不能把极少数蜕化变质的党员和党组织等同起来,他们并不能代表党。经过这几次认知意识的转变,我觉得入党前最重要的就是要正确和全面地认识我们的党。三次产生的入党动机,又三次被自己否定,我的思想仿佛又活跃不起来了,我怕再走错了方向,绕了弯路。此时,我的父亲对我的
分子动力学概述
分子动力学 分子动力学方法是一种计算机模拟实验方法,是研究凝聚态系统的有力工具。该技术不仅可以得到原子的运动轨迹,还可以观察到原子运动过程中各种微观细节。它是对理论计算和实验的有力补充。 分子动力学总是假定原子的运动服从某种确定的描述,这种描叙可以牛顿方程、拉格朗日方程或哈密顿方程所确定的描述,也就是说原子的运动和确定的轨迹联系在一起。在忽略核子的量子效应和Born-Oppenheimer绝热近似下,分子动力学的这一种假设是可行的[1]。所谓绝热近似也就是要求在分子动力学过程中的每一瞬间电子都处于原子结构的基态。要进行分子动力学模拟就必须知道原子间的相互作用势。在分子动力学模拟中,我们一般采用经验势来代替原子间的相互作用势,如Lennard-Jones势、Mores势、EAM原子嵌入势、F-S多体势。然而采用经验势必然丢失了局域电子结构之间存在的强相关作用信息,即不能得到原子动力学过程中的电子性质[1]。 事实上,分子动力学就是模拟原子系统的趋衡过程。实际上,分子动力学方法就是确定某一描述与初始条件、边值关系的数值解。我们假定系统经过M步长之后达到稳定,而这
一稳定状态正是我们所求的。 1、分子动力学的算法分析 首先,我们假定我们研究的系统服从 Newton 方程所确定的描述,即: )(1 )(.. t F m t r = (1) 式中r(t)表征原子在t 时刻的位置矢量 F(t)表征原子在t 时刻所受到的力,它与所有原子的位置矢有关 m 表征原子的质量。 如果我们给定初始条件,即方程(1)的定解条件r(0)和v(0),那么方程(1)的解就可以确定。60年代中期发展了大量的分子动力学算法,如两步差分算法[2]、预测-校正算法 [3] 、中心差分算法[4]、蛙跳算法[5]等等。为了方便导出它们, 我们以Euler 一步法[6] 来讨论之。我们令)()(.. t r t v =(表征粒子 的速度),则有: ) ()()(1 )()(... . t v t r t F m t r t v === (2) 记??? ? ??????=? ? ? ???=)()(1)()()()(. t v t F m t f t r t v t w (3)
分子动力学方法模拟基本步骤
分子动力学方法模拟基本步骤 1.第一步 即模型的设定,也就是势函数的选取。势函数的研究和物理系统上对物质的描述研究息息相关。最早是硬球势,即小于临界值时无穷大,大于等于临界值时为零。常用的是LJ势函数,还有EAM势函数,不同的物质状态描述用不同的势函数。 模型势函数一旦确定,就可以根据物理学规律求得模拟中的守恒量。 2 第二步 给定初始条件。运动方程的求解需要知道粒子的初始位置和速度,不同的算法要求不同的初始条件。如:verlet算法需要两组坐标来启动计算,一组零时刻的坐标,一组是前进一个时间步的坐标或者一组零时刻的速度值。 一般意思上讲系统的初始条件不可能知道,实际上也不需要精确选择代求系统的初始条件,因为模拟实践足够长时,系统就会忘掉初始条件。当然,合理的初始条件可以加快系统趋于平衡的时间和步伐,获得好的精度。 常用的初始条件可以选择为:令初始位置在差分划分网格的格子上,初始速度则从玻尔兹曼分布随机抽样得到;令初始位置随机的偏离差分划分网格的格子上,初始速度为零;令初始位置随机的偏离差分划分网格的格子上,初始速度也是从玻尔兹曼分布随机抽样得到。 第三步 趋于平衡计算。在边界条件和初始条件给定后就可以解运动方程,进行分子动力学模拟。但这样计算出的系统是不会具有所要求的系统的能量,并且这个状态本身也还不是一个平衡态。 为使得系统平衡,模拟中设计一个趋衡过程,即在这个过程中,我们增加或者从系统中移出能量,直到持续给出确定的能量值。我们称这时的系统已经达到平衡。这段达到平衡的时间成为驰豫时间。 分子动力学中,时间步长的大小选择十分重要,决定了模拟所需要的时间。为了减小误差,步长要小,但小了系统模拟的驰豫时间就长了。因此根据经验选择适当的步长。如,对一个具有几百个氩气Ar分子的体系,lj势函数,发现取h为0.01量级,可以得到很好的相图。这里选择的h是没有量纲的,实际上这样选择的h对应的时间在10-14s的量级呢。如果模拟1000步,系统达到平衡,驰豫时间只有10-11s。 第四步 宏观物理量的计算。实际计算宏观的物理量往往是在模拟的最后揭短进行的。它是沿相空间轨迹求平均来计算得到的(时间平均代替系综平均)
入党积极分子入党自我简介
入党积极分子入党自我简介 篇一 我叫xx,19xx年x月x日出生于xx市的一个普通家庭,xxxx 年加入共青团组织,并于xxxx年底递交了入党申请书,现在就读于xx大学xx系xx班。我志愿加入中国共产党,成为一名光荣的共产党员。因为中国共产党是中国工人阶级的先锋队,是中国各族人民利益的忠实代表,是中国社会主义事业的领导核心。党的最终目标是实现共产主义的社会制度。作为一名入党积极分子,首先要解决为什么要入党的问题。因为入党动机是激励我入党的主观原因,从根本上决定了作为一名党员应具有的素质和行为,也是个人世界观、人生观的集中反映。其实正确的入党动机的形成并不在一朝一夕,而是在我三年多的学习、思考中渐渐地体会到的。 下面我详细地就我的入党动机问题谈一下我的心历里程。刚递交入党申请书时,是因为看到别人写入党申请,如果自己不写,怕老师,同学说自己不要求进步,于是也随大流写份申请,但是其间的所写却都是从书上,从党员父亲那里抄来、学来的。其实那时的我对党缺乏真正的认识,尽管上过几次党课,但对党的基本知识还是知之甚少,思想上并没有迫切要求入党的愿望。这是一种对党、对个人都不认真、不严肃的政治态度。后来到高中毕业前夕,看到有的同学入了党,在同学中有一定的威望,羡慕不
已,于是自己赶紧写了几份思想汇报,企盼有一天自己能获此荣耀,得到精神上的满足。其实这是一种入党动机不纯的表现。入党不是给别人炫耀的,更不是用来满足自己虚荣心的,而是应该踏踏实实为党奉献,全心全意为人民服务,在学习和生活中发挥先锋模范作用。 大一时,在上党的基本知识概论课中学习了党的基本知识,同时有更多的时间和机会接触到身边的党员,政治视野也得到了扩充,看到当前存在党风不正的现象,希望自己能加入到党组织,做一名优秀的党员,重新确定党的形象。这种看法现在想起来是很片面和狭隘的,这种错误想法的产生究于对党的认识还太模糊,不能以因为有些党员存在着这样那样的缺点就否定他们是无产阶级的先锋战士,一直影响对党性的认识,也不能把极少数蜕化变质的党员和党组织等同起来,他们并不能代表党。经过这几次认知意识的转变,我觉得入党前最重要的就是要正确和全面地认识我们的党。三次产生的入党动机,又三次被自己否定,我的思想仿佛又活跃不起来了,我怕再走错了方向,绕了弯路。此时,我的父亲对我的教育给了我很大的启示。平日里,我最喜欢同父亲探讨我的思想动态,因为他不仅是最了解我的人,而且是个老党员。他对我反复地转变并不惊讶,反而给予了赞扬,他说,那是我在一次次为自己诠释对党的认识,是我确立正确的入党动机的“必修课”。他对我说,入党是自己的事,而且同学习一样,重要的是独立思考的能力,要会观察,会总结,会引申,要去看书学习,去思考,是要花一番气力的。有些人在入党之后一段时间,甚至是一生都要不断补充对入党的思考,也包括对入党动机的更
分子动力学
第五章 分子动力学 第一节 Verlet 算法 牛顿方程 i i i m f dt r d 2 2 记 N r r r R ,,21 N N m f m f m f G ,,221 1 方程写为 2 d R G dt v v 三点公式 2 42 111122n n n n n n n R R R G R R v v v v v v v r 如果给出初始条件0R 和1R ,可求解方程,但常常给出的初 始条件是00,v R , 那么 02 0012 G v R R (为什么? 因为dv G dt r ,所以,0000 ()'(')t v t v dt G t v t G r r v ;, 所以,210000000 '(')R R dt v t G R v G r r r r r ;) 方法的优点: 保持时间反演不变性,即令 n n , 方程形式 不变 (尽管误差会破坏这一对称性)
如果问题与v 无关,计算精度相当高 方法的缺点: n v v 必须用到1n R v (为什么是缺点?) 另一方案 2 221112! ()2 n n n n n n n n R R v G v v G G v v v v v 缺点:失去时间反演不变性 第二节 多体问题的基本方法 (阅读材料) 全同粒子,概率分布为 N r r r W R W 21, 物理量平均值 1i i A A R W R dR dR dr Z Z W R dR v v v v v v v 分子动力学 1 lim dt t A A n 个粒子处于 n r r ,1的分布密度函数 N n n n r d r d R W n N N Z r r r 121!!1, !! n N N 来自N 个粒子中取n 个的组合数 例如:N n 是1
分子动力学模拟
分子动力学模拟 分子动力学就是一门结合物理,数学与化学的综合技术。分子动力学就是一套分子模拟方法,该方法主要就是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量与其她宏观性质。 这门技术的发展进程就是: 1980年:恒压条件下的动力学方法(Andersenの方法、Parrinello-Rahman法) 1983年:非平衡态动力学方法(Gillan and Dixon) 1984年:恒温条件下的动力学方法(能势‐フーバーの方法) 1985年:第一原理分子动力学法(→カー?パリネロ法) 1991年:巨正则系综的分子动力学方法(Cagin and Pettit)、 最新的巨正则系综,即为组成系综的系统与一温度为T、化学势为μ的很大的热源、粒子源相接触,此时系统不仅同热源有能量交换,而且可以同粒子源有粒子的交换,最后达到平衡,这种系综称巨正则系综。 进行分子动力学模拟的第一步就是确定起始构型,一个能量较低的起始构型就是进行分子模拟的基础,一般分子的其实构型主要就是来自实验数据或量子化学计算。在确定起始构型之后要赋予构成分子的各个原子速度,这一速度就是根据玻尔兹曼分布随机生成,由于速度的分布符合玻尔兹曼统计,因此在这个阶段,体系的温度就是恒定的。另外,在随机生成各个原子的运动速度之后须进行调整,使得体系总体在各个方向上的动量之与为零,即保证体系没有平动位移。 由上一步确定的分子组建平衡相,在构建平衡相的时候会对构型、温度等参数加以监控。 进入生产相之后体系中的分子与分子中的原子开始根据初始速度运动,可以想象其间会发生吸引、排斥乃至碰撞,这时就根据牛顿力学与预先给定的粒子间相互作用势来对各个例子的运动轨迹进行计算,在这个过程中,体系总能量不变,但分子内部势能与动能不断相互转化,从而体系的温度也不断变化,在整个过程中,体系会遍历势能面上的各个点,计算的样本正就是在这个过程中抽取的。 用抽样所得体系的各个状态计算当时体系的势能,进而计算构型积分。 作用势的选择与动力学计算的关系极为密切,选择不同的作用势,体系的势能面会有不同的形状,动力学计算所得的分子运动与分子内部运动的轨迹也会不同,进而影响到抽样的结果与抽样结果的势能计算,在计算宏观体积与微观成分关系的时候主要采用刚球模型的二体势,计算系统能量,熵等关系时早期多采用Lennard-Jones、morse势等双体势模型,对于金属计算,主要采用morse势,但就是由于通过实验拟合的对势容易导致柯西关系,与实验不符,因此在后来的模拟中有人提出采用EAM等多体势模型,或者采用第一性原理计算结果通过一定的物理方法来拟合二体势函数。但就是对于二体势模型,多体势往往缺乏明确的表达式,参量很多,模拟收敛速度很慢,给应用带来很大困难,因此在一般应用中,通过第一性原理计算结果拟合势函数的L-J,morse等势模型的应用仍非常广泛。 分子动力学计算的基本思想就是赋予分子体系初始运动状态之后,利用分子的自然运动在相空间中抽取样本进行统计计算,时间步长就就是抽样的间隔,因而时间步长的选取对动力学模拟非常重要。太长的时间步长会造成分子间的激烈碰撞,体系数据溢出;太短的时间步长会降低模拟过程搜索相空间的能力,因此一般选取的时间步长为体系各个自由度中最短运动周期的十分之一。但就是通常情况下,体系各自由度中运动周期最短的就是各个化学键的振动,而这种运动对计算某些宏观性质并不产生影响,因此就产生了屏蔽分子内部振动或其她无关运动的约束动力学,约束动力学可以有效地增长分子动力学模拟时间步长,提高搜索相空间的能
分子动力学在材料科学中的应用
分子动力学在材料科学中的应用 摘要:本文综述了几种常见条件下的分子动力学模拟方法以及分子动力学模拟的最新发展趋势.介绍用分子动力学模拟方法研究固休的休相结构,表面问题,界面问题以及薄膜形成过程等方面的研究成果。 关键词:分子动力学; 计算机模拟; 材料科学 1引言 分子动力学(Molecular Dyanmica,简称MD)用于计算以固体、液体、气体为模型的单个分子运动,它是探索各种现象本质和某些新规律的一种强有力的计算机模拟方法,具有沟通宏观特性与微观结构的作用,对于许多在理论分析和实验观察上难以理解的现象可以做出一定的解释[1]。MD方法不要求模型过分简化,可以基于分子(原子、离子)的排列和运动的模拟结果直接计算求和以实现宏观现象中的数值估算。可以直接模拟许多宏观现象,取得和实验相符合或可以比较的结果,还可以提供微观结构、运动以及它们和体系宏观性质之间关系的极其明确的图象[2]。MD以其不带近似、跟踪粒子轨迹、模拟结果准确[3],而倍受研究者的关注,在物理、化学、材料、摩擦学等学科及纳米机械加工中得到广泛而成功的应用。本文主要评述MD方法在材料科学中的应用. 目前在材料微观结构的研究中,由于实验条件的限制,使得许多重要的微观结构的信息难以得到,如,对于由液态金属快速凝固的非晶转变过程,其微观结构的瞬时变化根本无法用实验仪器去测量。理论分析、实验测定及模拟计算已成为现代材料科学研究的3种主要方法[2]。20世纪90年代以来,由于计算机科学和技术的飞速发展,模拟计算的地位日渐突显。计算机模拟可以提供实验上尚无法获得或很难获得的信息。虽然计算机模拟不能完全取代实验,但可以用来指导实验的进行,从而促进理论和实践的发展,所以有必要对这一领域进行介绍。 2 分子动力学基本原理 分子动力学将连续介质看成由N个原子或分子组成的粒子系统,各粒子之间的作用力可以通过量子力学势能函数求导得出,忽略量子效应后,运用经典牛顿力学建立系统粒子运动数学模型,通过数值求解得到粒子在相空间的运动轨迹,然后由统计物理学原理得出该系统相应的宏观动态、静态特性。图1所示是MD
第六章分子动力学方法
第六章 分子动力学方法 6.1引言 对于一个多粒子体系的实验观测物理量的数值可以由总的平均得到。但是由于实验体系又非常大,我们不可能计算求得所有涉及到的态的物理量数值的总平均。按照产生位形变化的方法,我们有两类方法对有限的一系列态的物理量做统计平均: 第一类是随机模拟方法。它是实现Gibbs的统计力学途径。在此方法中,体系位形的转变是通过马尔科夫(Markov)过程,由随机性的演化引起的。这里的马尔科夫过程相当于是内禀动力学在概率方面的对应物。该方法可以被用到没有任何内禀动力学模型体系的模拟上。随机模拟方法计算的程序简单,占内存少,但是该方法难于处理非平衡态的问题。
另一类为确定性模拟方法,即统计物理中的所谓分子动力学方法(Molecular Dynamics Method)。这种方法广泛地用于研究经典的多粒子体系的研究中。该方法是按该体系内部的内禀动力学规律来计算并确定位形的转变。它首先需要建立一组分子的运动方程,并通过直接对系统中的一个个分子运动方程进行数值求解,得到每个时刻各个分子的坐标与动量,即在相空间的运动轨迹,再利用统计计算方法得到多体系统的静态和动态特性, 从而得到系统的宏观性质。因此,分子动力学模拟方法可以看作是体系在一段时间内的发展过程的模拟。在这样的处理过程中我们可以看出:分子动力学方法中不存在任何随机因素。 系统的动力学机制决定运动方程的形式: 在分子动力学方法处理过程中,方程组的建立是通过对物理体系的微观数学描述给出的。在这个微观的物理体系中,每个分子都各自服从经典的牛顿力学。每个分子运动的内禀动力学是用理论力学上的哈密顿量或者拉格朗日量来描述,也可以直接用牛顿运动方程来描述。这种方法可以处理与时间有关的过程,因而可以处理非平衡态问题。但是使用该方法的程序较复杂,计算量大,占内存也多。
vasp做分子动力学
vasp做分子动力学的好处,由于vasp是近些年开发的比较成熟的软件,在做电子scf速度方面有较好的优势。 缺点:可选系综太少。 尽管如此,对于大多数有关分子动力学的任务还是可以胜任的。 主要使用的系综是NVT和NVE。 下面我将对主要参数进行介绍! 一般做分子动力学的时候都需要较多原子,一般都超过100个。 当原子数多的时候,k点实际就需要较少了。有的时候用一个k点就行,不过这都需要严格的测试。通常超过200个原子的时候,用一个k点,即Gamma点就可以了。 INCAR: EDIFF 一般来说,用1E-4或者1E-5都可以,这个参数只是对第一个离子步的自洽影响大一些,对于长时间的分子动力学的模拟,精度小一点也无所谓,但不能太小。 IBRION=0 分子动力学模拟 IALGO=48 一般用48,对于原子数较多,这个优化方式较好。 NSW=1000 多少个时间步长。 POTIM=3 时间步长,单位fs,通常1到3. ISIF=2 计算外界的压力. NBLOCK= 1 多少个时间步长,写一次CONTCAR,CHG和CHGCAR,PCDAT. KBLOCK=50 NBLOCK*KBLOCK个步长写一次XDATCAR. ISMEAR=-1 费米迪拉克分布. SIGMA =0.05 单位:电子伏 NELMIN=8 一般用6到8,最小的电子scf数.太少的话,收敛的不好. LREAL=A APACO=10 径向分布函数距离,单位是埃. NPACO=200 径向分布函数插的点数. LCHARG=F 尽量不写电荷密度,否则CHG文件太大. TEBEG=300 初始温度. TEEND=300 终态温度。不设的话,等于TEBEG. SMASS -3 NVE ensemble;-1 用来做模拟退火;大于0 NVT 系综。 ///////////////////////////////////////////////////////////////////// ///////////////////////////////////////////////////////////////////// 1)收敛判据的选择 结构弛豫的判据一般有两种选择:能量和力。这两者是相关的,理想情况下,能量收敛到基态,力也应该是收敛到平衡态的。但是数值计算过程上的差异导致以二者为判据的收敛速度差异很大,力收敛速度绝大部分情况下都慢于能量收敛速度。这是因为力的计算是在能量的基础上进行的,能量对坐标的一阶导数得到力。计算量的增大和误差的传递导致力收敛慢。 到底是以能量为收敛判据,还是以力为收敛判据呢?关心能量的人,觉得以能量
水玻璃基本知识简介
硅酸钠基本知识简介 英文名:Sodium silicate, Water glass. 硅酸钠是无色固体,密度2.4g/cm3,熔点1321K(1088℃)。溶于水成粘稠溶液,俗称水玻璃、泡花碱。是一种无机粘合剂。 固体硅酸钠南方多称水玻璃,北方多称泡花碱,硅酸钠的水溶液通称水玻璃。纯固体硅酸钠为无色透明固体,市售硅酸钠多含有某些杂质,略带浅蓝色。 硅酸钠俗称水玻璃,液体硅酸钠为无色、略带色的透明或半透明粘稠状液体。固体硅酸钠为无色、略带色的透明或半透明玻璃块状体。形态分为液体、固体、水淬三种。理论上称这类物质为“胶体”。普通硅酸钠为略带浅蓝色块状或颗粒状固体,高温高压溶解后是略带色的透明或半透明粘稠液体。 市面上出售的AR分析纯水玻璃为Na2SiO3·9H2O,放置在空气中吸潮、结块。在水中的极易溶解。 泡花碱也就是硅酸钠(Na2SiO3),溶于水后形成的粘稠溶液,通称水玻璃,呈碱性。它的用途非常广泛,往往根据其粘结性强的特点,被用做硅胶,而且耐酸、耐热。有毒,但对一般的接触没有影响,误食则会对人体的肝脏造成危害 分类介绍 1、硅酸钠分两种,一种为偏硅酸钠,化学式Na2SiO3,式量122.00。另一种为正硅酸钠,化学式Na4SiO4,式量184.04。 2、正硅酸钠是无色晶体,熔点 1291K(1088℃),不多见。水玻璃溶液因水解而呈碱性(比纯碱稍强)。因系弱酸盐所以遇盐酸,硫酸、硝酸、二氧化碳都能析出硅酸。保存时应密切防止二氧化碳进入,并应使用橡胶塞以防粘住磨口玻璃塞。工业上常用纯碱与石英共熔制取Na2CO3+SiO2→Na2SiO3+CO2↑,制品常因含亚铁盐而带浅蓝绿色。用为无机粘接制剂(可与滑石粉等混合共用),肥皂填充剂,调制耐酸混凝土,加入颜料后可做外墙的涂料,灌入古建筑基础土壤中使土壤坚固以防倒塌。 3、偏硅酸钠是普通泡化碱与烧碱水热反应而制得的低分子晶体,商品有无水、五水和九水合物,其中九水合物只有我国市场上存在,是在上世纪80年代急需偏硅酸钠而仓促开发的技术含量较低的应急产品,因其熔点只有42℃,贮存时很容易变为液体或膏状,正逐步被淘汰,但由于一些用户习惯和一些领域对结晶水不是很在意,九水偏硅酸钠还是有一定市场。 生产方法 硅酸钠的生产方法分干法(固相法)和湿法(液相法)两种。
入党积极分子入党自我介绍
入党积极分子入党自我介绍 篇一 我叫×××,女,共青团员,汉族,籍贯××。现就读于××××大学建筑 工程学院××级工程管理1班。 我于××××年七月二十七日出生在一个普通而温暖的农村家庭中。我的父 母都是最普通的工人,父亲×××,是上海威捷制衣机械有限公司的机械工人, 谦虚严谨,细致谨慎的工作作风从小就深深地感染着我。母亲×××乐观开朗, 总是能给我无限动力。我的父母都是忠厚老实的群众,虽然他们并不是党员,但 却都深爱着我们的党和国家。就在这样一个普通而又温暖的家庭,我健康、幸福 地成长。成长环境使我对中国共产党有了由浅至深的认识,并逐步形成了共产主 义的世界观、人生观。 我是在党的教育下成长起来的。爷爷就是优秀共产党员,影响了我父亲和我。××年九月我进入小学,临学前父亲教育我,鲜艳的红领巾是先辈的鲜血染红的,是少先先锋队的标志,只有像解放军战士那样不怕苦,最勇敢的人才配戴上它。 我牢记父亲的话。在加入少先队戴红领巾之前我们要表现良好,那样表示你积极、进步;上小学一年级后,学习上,努力刻苦,争当先进;劳动中,不怕脏,不怕累。最后在小学二年级光荣地加入了中国少年先锋队,我抚摸着胸前的红领巾暗暗下 定决心,一定要更加进步,将来还要向团组织靠拢。 从此,我学习更加努力了,在班上学习一直很努力。××年X月我以年级第 三的名次从××中心小学毕业,进入××中学就读。随着知识的积累和年龄的增长,我在思想上逐渐懂得了,青年人要成长进步必须靠近团组织,主动接受团组 织的教育和培养。通过组织的帮助和自己的努力,于××年X月的时候,我光荣
地加入了中国共产主义青年团。中国共青团是中国先进青年的群众组织,是中国 共产党的得力助手和后备军。当我在团旗下举起右手庄严地宣誓时,心潮澎湃!我 暗下决心:一定要好好学习,全面发展,在各方面都要起模范带头作用,把自己培 养成为跨世纪的社会主义建设者和接班人,为我国的社会主义现代化建设贡献自 己的全部力量。在这一思想指导下,我刻苦学习政治理论和科学文化知识,学习 成绩优秀,是班上的学习委员,在此期间在学校连续三年获得校三好学生,优秀 团员以及优秀干部的称号。初三毕业时,我取得推优生资格,保送进××市实验性、示范性高中之一--××中学,并取得了606.5分的××区前几名的中考成绩。 ××年X月入学时,我就读于××中学重点班,翻开了我人生征程崭新的一页,我朝着新的目标开始了新的奋斗。在校期间,我虚心学习,端正自己的学习 态度,在高一和高二均是班中的学习委员。 经过一年的高三奋斗,××年我参加高考,考进了××××大学,翻开了我 人生征程崭新的一页,我对着新的目标开始了新的拼搏。入学不久,我就怀着十 分激动的心情向党组织递交了入党申请书,从此我抱着为共产主义事业奋斗终生 的决心,时时处处以党员的标准要求自己,我踏踏实实的工作学习,经常为同学 做一些力所能及的事,关心同学们的学习和生活,在各方面起到了表率作用。 在大学期间,在思想上,我积极要求上进,人只有树立正确的人生观,树立 远大理想,无止境地追求,才会生活得更有意义。我在担任学生会担任干事期间,认真履行自己的职责,对学生管理事务注入了很大的热情,而且坚持锻炼自己做 好学生会工作,学好专业课程两方面的能力,就在前不久我以优异的成绩拿到了 二等奖学金,在思想上积极进取,努力向党组织靠拢,认真学习马克思列宁主义 毛泽东思想,党的章程,对党的认识逐渐清晰,同时我也知道,我对党的认识仍 比较肤浅,需要不断学习与锻炼来提高自己。
入党积极分子个人简历
入党积极分子个人简历 我叫xxx,今年18岁,是历史学师范专业的一名学生,女,汉族,出生于重庆市的一个普通工人家庭,现任学习委员。 1998年9月1日,我怀着童年的梦想上了重庆市xx区xx小学。接触的一切都是那么陌生、新奇,久而久之,又是那么的熟悉。在二年级,我早早埋藏在心中的愿望实现了,终于戴上了革命先烈用鲜血染成的红旗一角——红领巾。从那以后,我担任了班级劳动委员、少先队中队长的职务,由于表现突出,被评为优秀少先队员、三好学生等。在老师的培养下,加上我自身的努力,2004年,我以优异的成绩考入当地的xx中学。在初中一年级时终于盼来了期待已久的入团宣誓!中国共青团是中国先进青年的群众组织,是中国共产党的得力助手和后备军。中国共产主义青年团是广大青年在实践中学习共产主义的学校,在那里我加强了对党的认识。当我在团旗下举起右手庄严地宣誓时,心潮澎湃!我清楚地知道团员这个光荣的称号也意味着责任,从此我处处以团员标准要求自己。我告诫自己要做一个党的好孩子,并且时刻督促着我自己一定要戒骄戒躁,要继续努力,将来向党组织靠拢。期间获得老师和同学的一致好评,还被评“学习积极分子”等荣誉称号。 高中是青年学生世界观形成和确立的重要阶段。我觉得在高中期间认真学习文化知识固然重要,但政治学习也不能忽视。我生长在党员之家,在祖父的熏陶下,我利用课余时间学习、翻阅了一些近代历
史与政治,这使我对共产主义有了初步认识,再联系平常在报纸上、电视中看到的共产党员们光明磊落、无私奉献的精神与舍己救人的英雄事迹后,在我心中泛起了波澜…… 作为一名入党积极分子,我明白以实际行动争取入党,必须持之以恒,从申请入党的那天起,就应该以正确的态度和真诚的努力争取早日成为一名名副其实的共产党员。作为一个积极争取入党的人,我不仅要做一名合格的大学生,还应该是对党的路线方针、政策的关心者和拥护者。在日常生活中我积极关心时事政治,尤其是关于党制定的纲领路线、方针政策的消息和评论。 理想是远大的,但还要从实处入手,我会从现在开始以实际行动开始努力,以一个党员的标准,严格要求自己,尽量缩小与党员标准之间的差距,如果我被党组织吸收,我会更加坚定共产主义理想信念,遵守党的章程。以合格党员的标准时刻要求自己,积极发挥党员的先锋模范作用。自觉接受党的教导,按照“三个代表”的要求贯彻落实党的路线、方针和政策,不断把建设有中国特色的社会主义事业推向前进。
【学术讲坛】分子动力学介绍
【专业】计算物理 【研究方向】分子动力学模拟 【学术讲坛】 1、分子动力学简介: 分子动力学方法是一种计算机模拟实验方法,是研究凝聚态系统的有力工具。该技术不仅可以得到原子的运动轨迹,还可以观察到原子运动过程中各种微观细节。它是对理论计算和实验的有力补充。广泛应用于材料科学、生物物理和药物设计等。经典MD模拟,其系统规模在一般的计算机上也可达到数万个原子,模拟时间为纳秒量级。2006年进行了三千二百亿个原子的模拟(IBM lueGene/L)。 分子动力学总是假定原子的运动服从某种确定的描述,这种描叙可以牛顿方程、拉格朗日方程或哈密顿方程所确定的描述,也就是说原子的运动和确定的轨迹联系在一起。在忽略核子的量子效应和Born-Oppenheimer绝热近似下,分子动力学的这一种假设是可行的。所谓绝热近似也就是要求在分子动力学过程中的每一瞬间电子都处于原子结构的基态。要进行分子动力学模拟就必须知道原子间的相互作用势。 在分子动力学模拟中,我们一般采用经验势来代替原子间的相互作用势,如 Lennard-Jones势、Mores势、EAM原子嵌入势、F-S多体势。然而采用经验势必然丢失了局域电子结构之间存在的强相关作用信息,即不能得到原子动力学过程中的电子性质。 详细介绍请见附件。 2、分子模拟的三步法和大致分类 三步法: 第一步:建模。包括几何建模,物理建模,化学建模,力学建模。初始条件的设定,这里要从微观和宏观两个方面进行考虑。 第二步:过程。这里就是体现所谓分子动力学特点的地方。包括对运动方程的积分的有效算法。对实际的过程的模拟算法。关键是分清楚平衡和非平衡,静态和动态以及准静态情况。 第三步:分析。这里是做学问的关键。你需要从以上的计算的结果中提取年需要的特征,说明你的问题的实质和结果。因此关键是统计、平均、定义、计算。比如温度、体积、压力、应力等宏观量和微观过程量是怎么联系的。 有了这三步,你就可以做一个好的分子动力学专家了。推而广之,其实所谓的介观模拟,蒙特卡罗模拟、有限元模拟都是一个道理。
分子动力学模拟及其在材料中的研究进展汇总
《材料计算设计基础》 学号: 流水号: 姓名: 完成日期:
分子动力学模拟及其在材料中的研究进展 摘要:本文综述了分子动力学模拟技术的发展,介绍了分子动力学的分类、运动方程的求解、初始条件和边界条件的选取、平衡系综及其控制、感兴趣量的提取以及分子动力学模拟在材料中的研究进展。 关键词:分子动力学模拟平衡态系综金属材料感兴趣量径向分布函数 引言 科学工作者在长期的科学研究实践中发现,当实验研究方法不能满足研究工作的需求时,用计算机模拟却可以提供实验上尚无法获得或很难获得的重要信息;尽管计算机模拟不能完全取代实验,但可以用来指导实验,并验证某些理论假设,从而促进理论和实验的发展。特别是在材料形成过程中许多与原子有关的微观细节,在实验中基本上是无法获得的,而在计算机模拟中即可以方便地得到。这种优点使分子动力学模拟在金属材料研究中显得非常有吸引力。 分子动力学MD (Molecular Dynamics)模拟就是用计算机方法来表示统计力学,作为实验的一个辅助手段。MD模拟就是对于原子核和电子所构成的多体系统,求解运动方程(如牛顿方程、哈密顿方程或拉格朗日方程),其中每一个原子核被视为在全部其它原子核和电子作用下运动,通过分析系统中各粒子的受力情况,用经典或量子的方法求解系统中各粒子在某时刻的位置和速度,以确定粒子的运动状态,进而计算系统的结构和性质。该模拟技术主要涉及粒子运动的动力学问题,与蒙特卡罗模拟方法(简称MC)相比,分子动力学是一种“确定性方法”, 它所计算的是时间平均,而MC进行的是系综平均。然而按照统计力学各态历经假设,时间平均等价于系综平均。因此,两种方法严格的比较计算能给出几乎相同的结果。 经典的分子动力学方法是Alder等于1957年提出并首先在“硬球”液体模型下应用,发现了由Kirkwood在1939年根据统计力学预言的“刚性球组成的集合系统会发生有液相到结晶相的转变”。后来人们称这种相变为Alder相变。Rahman