PCR扩增的原理和操作步骤

PCR扩增反应的操作
第一节PCR扩增反应的基本原理
一、聚合酶链式反应(PCR)的基本构成
PCR是聚合酶链式反应的简称,指在引物指导下由酶催化的对特定模板(克隆或基因组DNA)的扩增反应,是模拟体内DNA复制过程,在体外特异性扩增DNA片段的一种技术,在分子生物学中有广泛的应用,包括用于DNA作图、DNA测序、分子系统遗传学等。
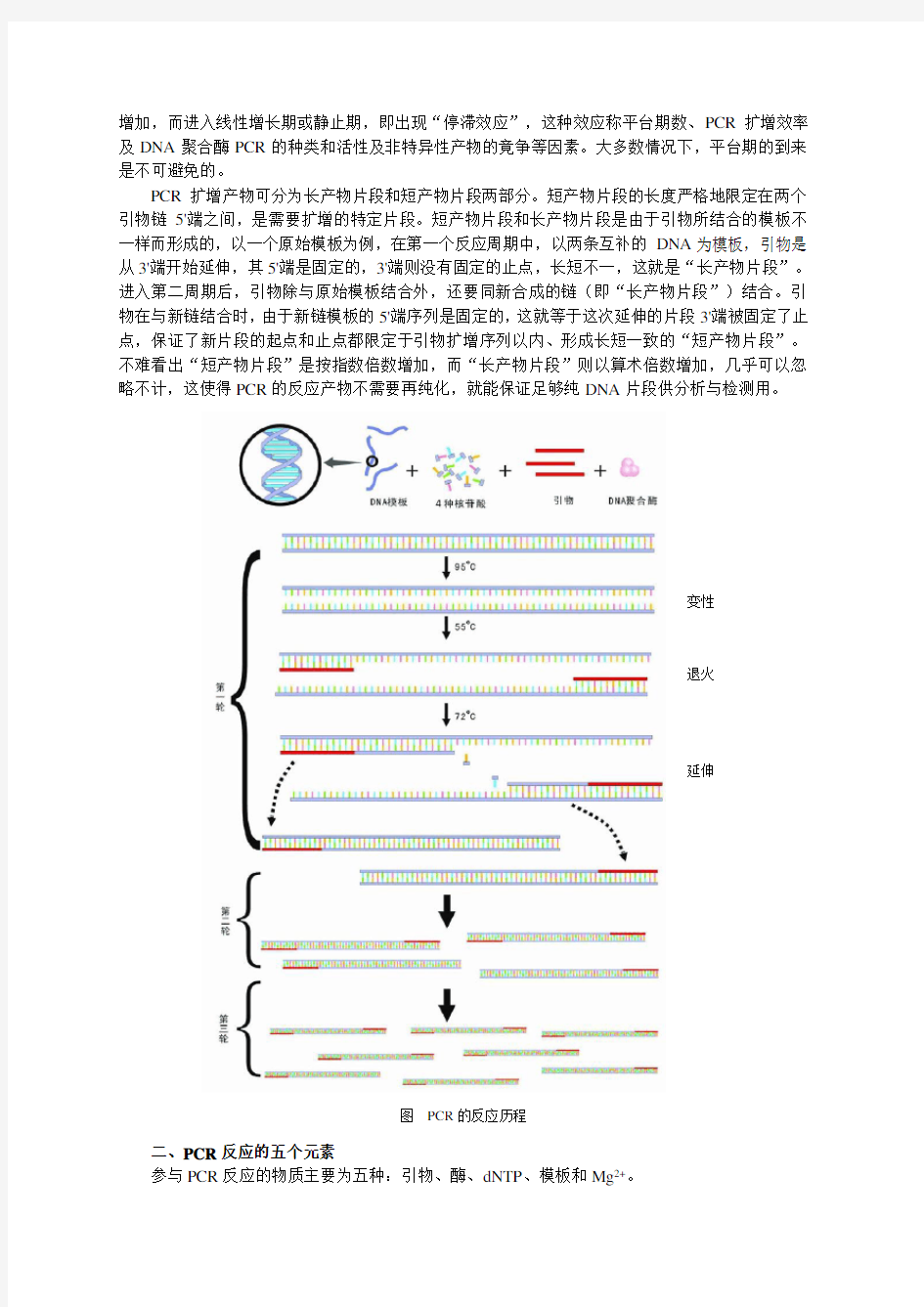
PCR基本原理是以单链DNA为模板,4种dNTP为底物,在模板3’末端有引物存在的情况下,用酶进行互补链的延伸,多次反复的循环能使微量的模板DNA得到极大程度的扩增。在微量离心管中,加入与待扩增的DNA片段两端已知序列分别互补的两个引物、适量的缓冲液、微量的DNA 膜板、四种dNTP溶液、耐热Taq DNA聚合酶、Mg2+等。反应时先将上述溶液加热,使模板DNA 在高温下变性,双链解开为单链状态;然后降低溶液温度,使合成引物在低温下与其靶序列配对,形成部分双链,称为退火;再将温度升至合适温度,在Taq DNA聚合酶的催化下,以dNTP为原料,引物沿5’→3’方向延伸,形成新的DNA片段,该片段又可作为下一轮反应的模板,如此重复改变温度,由高温变性、低温复性和适温延伸组成一个周期,反复循环,使目的基因得以迅速扩增。因此PCR循环过程为三部分构成:模板变性、引物退火、热稳定DNA聚合酶在适当温度下催化DNA链延伸合成(见图)。
1.模板DNA的变性
模板DNA加热到90~95℃时,双螺旋结构的氢键断裂,双链解开成为单链,称为DNA的变性,以便它与引物结合,为下轮反应作准备。变性温度与DNA中G-C含量有关,G-C间由三个氢键连接,而A-T间只有两个氢键相连,所以G-C含量较高的模板,其解链温度相对要高些。故PCR 中DNA变性需要的温度和时间与模板DNA的二级结构的复杂性、G-C含量高低等均有关。对于高G-C含量的模板DNA在实验中需添加一定量二甲基亚砜(DMSO),并且在PCR循环中起始阶段热变性温度可以采用97℃,时间适当延长,即所谓的热启动。
2.模板DNA与引物的退火
将反应混合物温度降低至37~65℃时,寡核苷酸引物与单链模板杂交,形成DNA模板-引物复合物。退火所需要的温度和时间取决于引物与靶序列的同源性程度及寡核苷酸的碱基组成。一般要求引物的浓度大大高于模板DNA的浓度,并由于引物的长度显着短于模板的长度,因此在退火时,引物与模板中的互补序列的配对速度比模板之间重新配对成双链的速度要快得多,退火时间一般为1~2min。
3.引物的延伸
DNA模板-引物复合物在Taq DNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条与模板DNA链互补的新链。重复循环变性-退火-延伸三过程,就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。延伸所需要的时间取决于模板DNA的长度。在72℃条件下,Taq DNA聚合酶催化的合成速度大约为40~60个碱基/秒。经过一轮“变性-退火-延伸”循环,模板拷贝数增加了一倍。在以后的循环中,新合成的DNA都可以起模板作用,因此每一轮循环以后,DNA拷贝数就增加一倍。每完成一个循环需2~4min,一次PCR经过30~40次循环,约2~3h。扩增初期,扩增的量呈直线上升,但是当引物、模板、聚合酶达到一定比值时,酶的催化反应趋于饱和,便出现所谓的“平台效应”,即靶DNA产物的浓度不再增加。
PCR的三个反应步骤反复进行,使DNA扩增量呈指数上升。反应最终的DNA扩增量可用Y =(1+X)n计算。Y代表DNA片段扩增后的拷贝数,X表示平(Y)均每次的扩增效率,n代表循环次数。平均扩增效率的理论值为100%,但在实际反应中平均效率达不到理论值。反应初期,靶序列DNA片段的增加呈指数形式,随着PCR产物的逐渐积累,被扩增的DNA片段不再呈指数
增加,而进入线性增长期或静止期,即出现“停滞效应”,这种效应称平台期数、PCR扩增效率及DNA聚合酶PCR的种类和活性及非特异性产物的竟争等因素。大多数情况下,平台期的到来是不可避免的。
PCR扩增产物可分为长产物片段和短产物片段两部分。短产物片段的长度严格地限定在两个引物链5'端之间,是需要扩增的特定片段。短产物片段和长产物片段是由于引物所结合的模板不一样而形成的,以一个原始模板为例,在第一个反应周期中,以两条互补的DNA为模板,引物是从3'端开始延伸,其5'端是固定的,3'端则没有固定的止点,长短不一,这就是“长产物片段”。进入第二周期后,引物除与原始模板结合外,还要同新合成的链(即“长产物片段”)结合。引物在与新链结合时,由于新链模板的5'端序列是固定的,这就等于这次延伸的片段3'端被固定了止点,保证了新片段的起点和止点都限定于引物扩增序列以内、形成长短一致的“短产物片段”。不难看出“短产物片段”是按指数倍数增加,而“长产物片段”则以算术倍数增加,几乎可以忽略不计,这使得PCR的反应产物不需要再纯化,就能保证足够纯DNA片段供分析与检测用。
图PCR的反应历程
二、PCR反应的五个元素
参与PCR反应的物质主要为五种:引物、酶、dNTP、模板和Mg2+。变性退火延伸
1.引物
引物是PCR特异性反应的关键,PCR产物的特异性取决于引物与模板DNA互补的程度。理论上,只要知道任何一段模板DNA序列,就能按其设计互补的寡核苷酸链做引物,利用PCR就可将模板DNA在体外大量扩增。引物设计有3条基本原则:首先引物与模板的序列要紧密互补,其次引物与引物之间避免形成稳定的二聚体或发夹结构,再次引物不能在模板的非目的位点引发DNA聚合反应(即错配)。
引物的选择将决定PCR产物的大小、位置、以及扩增区域的Tm值这个和扩增物产量有关的重要物理参数。好的引物设计可以避免背景和非特异产物的产生,甚至在RNA-PCR中也能识别cDNA或基因组模板。引物设计也极大的影响扩增产量:若使用设计粗糙的引物,产物将很少甚至没有;而使用正确设计的引物得到的产物量可接近于反应指数期的产量理论值。当然,即使有了好的引物,依然需要进行反应条件的优化,比如调整Mg2+浓度,使用特殊的共溶剂如二甲基亚砜、甲酰胺和甘油。对引物的设计不可能有一种包罗万象的规则确保PCR的成功,但遵循某些原则,则有助于引物的设计。
(1)引物长度
PCR特异性一般通过引物长度和退火温度来控制。引物的长度一般为15-30bp,常用的是18~27bp,但不应大于38bp。引物过短时会造成Tm值过低,在酶反应温度时不能与模板很好的配对;引物过长时又会造成Tm值过高,超过酶反应的最适温度,还会导致其延伸温度大于74℃,不适于Taq DNA聚合酶进行反应,而且合成长引物还会大大增加合成费用。
(2)引物碱基构成
引物的G+C含量以40~60%为宜,过高或过低都不利于引发反应,上下游引物的GC含量不能相差太大。其Tm值是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度,有效启动温度,一般高于Tm值5~10℃。若按公式Tm=4(G+C)+2(A+T)估计引物的Tm值,则有效引物的Tm为55~80℃,其Tm值最好接近72℃以使复性条件最佳。引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。尤其3′端不应超过3个连续的G或C,因这样会使引物在G+C富集序列区错误引发。
(3)引物二级结构
引物二级结构包括引物自身二聚体、发卡结构、引物间二聚体等。这些因素会影响引物和模板的结合从而影响引物效率。对于引物的3’末端形成的二聚体,应控制其ΔG大于-5.0 kcal/mol或少于三个连续的碱基互补,因为此种情形的引物二聚体有进一步形成更稳定结构的可能性,引物中间或5’端的要求可适当放宽。引物自身形成的发卡结构,也以3’端或近3’端对引物-模板结合影响更大;影响发卡结构的稳定性的因素除了碱基互补配对的键能之外,与茎环结构形式亦有很大的关系。应尽量避免3’末端有发卡结构的引物。
(4)引物3’端序列
引物3’末端和模板的碱基完全配对对于获得好的结果是非常重要的,而引物3’末端最后5到6个核苷酸的错配应尽可能的少。如果3’末端的错配过多,通过降低反应的退火温度来补偿这种错配不会有什么效果,反应几乎注定要失败。
引物3’末端的另一个问题是防止一对引物内的同源性。应特别注意引物不能互补,尤其是在3’末端。引物间的互补将导致不想要的引物双链体的出现,这样获得的PCR产物其实是引物自身的扩增。这将会在引物双链体产物和天然模板之间产生竞争PCR状态,从而影响扩增成功。
引物3’末端的稳定性由引物3’末端的碱基组成决定,一般考虑末端5个碱基的ΔG。?G值是指DNA双链形成所需的自由能,该值反映了双链结构内部碱基对的相对稳定性,此值的大小对扩增有较大的影响。应当选用3’端?G值较低(绝对值不超过9),负值大,则3’末端稳定性高,扩增效率更高。引物的3’端的?G值过高,容易在错配位点形成双链结构并引发DNA聚合反应。
需要注意的是,如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增特异性与效率。另外末位碱基为A的错配效率明显高于其他3个碱基,因此应当避免在引物的3’端使用碱基A。
(5)引物的5′端
引物的5′端限定着PCR产物的长度,它对扩增特异性影响不大。因此,可以被修饰而不影响扩增的特异性。引物5′端修饰包括:加酶切位点;标记生物素、荧光、地高辛、Eu3+等;引入蛋白质结合DNA序列;引入突变位点、插入与缺失突变序列和引入一启动子序列等。对于引入一至两个酶切位点,应在后续方案设计完毕后确定,便于后期的克隆实验,特别是在用于表达研究的目的基因的克隆工作中。
(6)引物的特异性
引物与非特异扩增序列的同源性不要超过70%或有连续8个互补碱基同源,特别是与待扩增的模板DNA之间要没有明显的相似序列。
2.酶及其浓度
Taq DNA多聚酶是耐热DNA聚合酶,是从水生栖热菌(Thermus aquaticus)中分离的。Taq DNA 聚合酶是一个单亚基,分子量为94 000 Da。具有5’-->3的聚合酶活力,5’-->3’的外切核酸酶活力,无3’-->5’的外切核酸酶活力,会在3’末端不依赖模板加入1个脱氧核苷酸(通常为A,故PCR产物克隆中有与之匹配的T载体),在体外实验中,Taq DNA聚合酶的出错率为10-4~10-5。此酶的发现使PCR广泛的被应用。
此酶具有以下特点:
(1)耐高温,在70℃下反应2小时后其残留活性在90%以上,在93℃下反应2小时后其残留活性是仍能保持60%,而在95℃下反应2小时后为原来的40%。
(2)在热变性时不会被钝化,故不必在扩增反应的每轮循环完成后再加新酶。
(3)一般扩增的PCR产物长度可达2.0 kb,且特异性也较高。
PCR的广泛应用得益于此酶,目前各试剂公司中开发了多种类型的Taq酶,有用于长片段扩增的酶,扩增长度极端可达40kb;有在常温条件下即可应用的常温PCR聚合酶;还有针对不同实验对象的酶等。
一典型的PCR反应约需的酶量为2.5u(总反应体积为50?l时),浓度过高可引起非特异性扩增,浓度过低则合成产物量减少。
3.dNTP的质量与浓度
dNTP的质量与浓度和PCR扩增效率有密切关系,dNTP粉呈颗粒状,如保存不当易变性失去生物学活性。dNTP溶液呈酸性,使用时应配成高浓度后,以1M NaOH或1M Tris。HCl的缓冲液将其pH调节到7.0~7.5,小量分装,-20℃冰冻保存。多次冻融会使dNTP降解。在PCR反应中,dNTP应为50~200?mol/l,尤其是注意4种dNTP的浓度要相等(等摩尔配制),如其中任何一种浓度不同于其它几种时(偏高或偏低),就会引起错配。浓度过低又会降低PCR产物的产量。dNTP 能与Mg2+结合,使游离的Mg2+浓度降低。
4.模板(靶基因)核酸
模板核酸的量与纯化程度,是PCR成败与否的关键环节之一,传统的DNA纯化方法通常采用SDS和蛋白酶K来消化处理标本。SDS的主要功能是:溶解细胞膜上的脂类与蛋白质,因而溶解膜蛋白而破坏细胞膜,并解离细胞中的核蛋白,SDS还能与蛋白质结合而沉淀;蛋白酶K能水解消化蛋白质,特别是与DNA结合的组蛋白,再用有机溶剂(酚与氯仿抽)抽提除去蛋白质和其它细胞组份,用乙醇或异丙醇沉淀核酸,该核酸即可作为模板用于PCR反应。一般临床检测标本,可采用快速简便的方法溶解细胞,裂解病原体,消化除去染色体的蛋白质使靶基因游离,直接用于PCR扩增。
模板DNA投入量对于细菌基因组DNA一般在1~10ng/?l,实验中模板浓度常常需要优化,一般可选择几个浓度梯度(浓度差以10倍为一个梯度)。在PCR反应中,过高的模板投入量往往会导致PCR实验的失败。
5.Mg2+浓度
Mg2+对PCR扩增的特异性和产量有显着的影响,在一般的PCR反应中,各种dNTP浓度为200?mol/l时,Mg2+浓度为1.5~2.0mmol/l为宜。Mg2+浓度过高,反应特异性降低,出现非特异扩增,浓度过低会降低Taq DNA聚合酶的活性,使反应产物减少。一般厂商提供的Taq DNA聚合酶均有相应的缓冲液,而Mg2+也已添加,如果特殊实验应采用无Mg2+的缓冲液,,在PCR反应体系中添加一定量的Mg2+。
三、PCR反应特点
PCR反应具有以下特点:
1.强特异性
PCR反应的特异性决定因素为:(1)引物与模板DNA特异性的结合;(2)碱基配对原则;(3)Taq DNA聚合酶合成反应的忠实性;(4)靶基因的特异性与保守性。
其中引物与模板的正确结合是关键,引物与模板的结合及引物链的延伸是遵循碱基配对原则的。聚合酶合成反应的忠实性及Taq DNA聚合酶耐高温性,使反应中模板与引物的结合(复性)可以在较高的温度下进行,结合的特异性大大增加,被扩增的靶基因片段也就能保持很高的正确度。再通过选择特异性和保守性高的靶基因区,其特异性程度就更高。
2.高灵敏度
PCR产物的生成量是以指数方式增加的,能将皮克(pg=10-12g)量级的起始待测模板扩增到微克(?g = 10-6g)水平。能从100万个细胞中检出一个靶细胞;在病毒的检测中,PCR的灵敏度可达3个RFU(空斑形成单位);在细菌学中最小检出率为3个细菌。
3.快速简便
PCR反应用耐高温的Taq DNA聚合酶,一次性地将反应液加好后,即在PCR仪上进行变性-退火-延伸反应,反应一般在2~4小时完成。扩增产物常用电泳分析,操作简单易推广,如采用特殊PCR仪(荧光时时定量PCR仪)则可全程监测PCR反应的结果,故耗时将更短。
4.低纯度模板
不需要分离病毒或细菌及培养细胞,DNA粗制品及总RNA等均可作为扩增模板。可直接用临床标本如血液、体腔液、洗嗽液、毛发、细胞、活组织等粗制的DNA扩增检测。
第二节PCR扩增反应的实施
一、PCR反应的条件
PCR反应条件为温度、时间和循环次数。
1.温度与时间的设置
基于PCR原理三步骤而设置变性-退火-延伸三个温度点。在标准反应中采用三温度点法,双链DNA在90~95℃变性,再迅速冷却至40~60℃,引物退火并结合到靶序列上,然后快速升温至70~75℃,在Taq DNA 聚合酶的作用下,使引物链沿模板延伸。对于较短靶基因(长度为100~300bp时)可采用二温度点法,除变性温度外、退火与延伸温度可合二为一,一般采用94℃变性,65℃左右退火与延伸(此温度Taq DNA酶仍有较高的催化活性)。
(1)变性温度与时间:变性温度低,解链不完全是导致PCR失败的最主要原因。一般情况下,93℃~94℃lmin足以使模板DNA变性,若低于93℃则需延长时间,但温度不能过高,因为高温环境对酶的活性有影响。此步若不能使靶基因模板或PCR产物完全变性,就会导致PCR失败。
(2)退火(复性)温度与时间:退火温度是影响PCR特异性的较重要因素。变性后温度快速冷却至40℃~60℃,可使引物和模板发生结合。由于模板DNA比引物复杂得多,引物和模板之间的碰撞结合机会远远高于模板互补链之间的碰撞。退火温度与时间,取决于引物的长度、碱
基组成及其浓度,还有靶基序列的长度。对于20个核苷酸,G+C含量约50%的引物,55℃为选择最适退火温度的起点较为理想。引物的复性温度可通过以下公式帮助选择合适的温度:Tm值(解链温度)=4(G+C)+2(A+T)
复性温度=Tm值-(5~10℃)
在Tm值允许范围内,选择较高的复性温度可大大减少引物和模板间的非特异性结合,提高P CR反应的特异性。复性时间一般为30~60sec,足以使引物与模板之间完全结合。
(3)延伸温度与时间:Taq DNA聚合酶的生物学活性:70~80℃,150核苷酸/S/酶分子;7 0℃,60核苷酸/S/酶分子;55℃,24核苷酸/S/酶分子;高于90℃时,DNA合成几乎不能进行。
(4)PCR反应的延伸温度一般选择在70~75℃之间,常用温度为72℃,过高的延伸温度不利于引物和模板的结合。PCR延伸反应的时间,可根据待扩增片段的长度而定,一般1kb以内的DNA片段,延伸时间1min是足够的。3~4kb的靶序列需3~4min;扩增10kb需延伸至15min。延伸进间过长会导致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。
2.循环次数
循环次数决定PCR扩增程度。PCR循环次数主要取决于模板DNA的浓度,一般的循环次数选在30~40次之间,循环次数越多,非特异性产物的量亦随之增多。
二、PCR扩增产物分析
PCR产物是否为特异性扩增,其结果是否准确可靠,必须对其进行严格的分析与鉴定,才能得出正确的结论。PCR产物的分析,可依据研究对象和目的不同而采用不同的分析方法。
1.凝胶电泳分析:PCR产物电泳,EB溴乙锭染色紫外仪下观察,初步判断产物的特异性。P CR产物片段的大小应与预计的一致,特别是多重PCR,应用多对引物,其产物片断都应符合预讦的大小,这是起码条件。
(1)琼脂糖凝胶电泳:通常应用1~2%的琼脂糖凝胶,供检测用。
(2)聚丙烯酰胺凝胶电泳:6~10%聚丙烯酰胺凝胶电泳分离效果比琼脂糖好,条带比较集中,可用于科研及检测分析。
2.酶切分析:根据PCR产物中限制性内切酶的位点,用相应的酶切、电泳分离后,获得符合理论的片段,此法既能进行产物的鉴定,又能对靶基因分型,还能进行变异性研究。
3.分子杂交:分子杂交是检测PCR产物特异性的有力证据,也是检测PCR 产物碱基突变的有效方法。
4.Southern印迹杂交:在两引物之间另合成一条寡核苷酸链(内部寡核苷酸)标记后做探针,与PCR产物杂交。此法既可作特异性鉴定,又可以提高检测PCR产物的灵敏度,还可知其分子量及条带形状,主要用于科研。
5.斑点杂交:将PCR产物点在硝酸纤维素膜或尼膜薄膜上,再用内部寡核苷酸探针杂交,观察有无着色斑点,主要用于PCR产物特异性鉴定及变异分析。
6.核酸序列分析:是检测PCR产物特异性的最可靠方法。
三、PCR结果异常分析
PCR产物的电泳检测时间一般为48h以内,有些最好于当日电泳检测,大于48h后带型不规则甚致消失。
1.假阴性,不出现扩增条带
PCR反应的关键环节有:①模板核酸的制备;②引物的质量与特异性;③酶的质量及活性;
④PCR循环条件。寻找原因亦应针对上述环节进行分析研究。
(1)模板:①模板中含有杂蛋白质;②模板中含有Taq酶抑制剂;③模板中蛋白质没有消化除净,特别是染色体中的组蛋白;④在提取制备模板时丢失过多,或吸入酚;⑤模板核酸变性不彻底。在酶和引物质量好时,不出现扩增带,极有可能是标本的消化处理,模板核酸提取过程出了毛病,因而要配制有效而稳定的消化处理液,其程序亦应固定不宜随意更改。
(2)酶失活:需更换新酶,或新旧两种酶同时使用,以分析是否因酶的活性丧失或不够而导致假阴性。需注意的是有时忘加Taq酶或溴乙锭。
(3)引物:引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不理想、容易弥散的常见原因。有些批号的引物合成质量有问题,两条引物一条浓度高,一条浓度低,造成低效率的不对称扩增,对策为:
①选定一个好的引物合成单位。
②引物的浓度不仅要看OD值,更要注重引物原液做琼脂糖凝胶电泳,一定要有引物条带出现,而且两引物带的亮度应大体一致,如一条引物有条带,一条引物无条带,此时做PCR有可能失败,应和引物合成单位协商解决。如一条引物亮度高,一条亮度低,在稀释引物时要平衡其浓度。
③引物应高浓度小量分装保存,防止多次冻融或长期放冰箱冷藏部分,导致引物变质降解失效。④引物设计不合理,如引物长度不够,引物之间形成二聚体等。
(4)Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。
(5)反应体积的改变:通常进行PCR扩增采用的体积为20?l、30?l、50?l或100?l,用多大体积进行PCR扩增,是根据科研和临床检测不同目的而设定,在做小体积如20?l后,再做大体积时,一定要模索条件,否则容易失败。
(6)物理原因:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。有时还有必要用标准的温度计,检测一下扩增仪或水溶锅内的变性、退火和延伸温度,这也是PCR失败的原因之一。
(7)靶序列变异:如靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。
2.假阳性
出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。可能的原因是:(1)引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。靶序列太短或引物太短,容易出现假阳性。需重新设计引物。
(2)靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。这种假阳性可用以下方法解决:①操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。②除酶及不能耐高温的物质外,所有试剂或器材均应高压消毒。所用离心管及样进枪头等均应一次性使用。③必要时,在加标本前,反应管和试剂用紫外线照射,以破坏存在的核酸。二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。
3.出现非特异性扩增带
PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。其次是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶量过多有时也会出现非特异性扩增。其对策有:①必要时重新设计引物。②减低酶量或调换另一来源的酶。③降低引物量,适当增加模板量,减少循环次数。④适当提高退火温度或采用二温度点法(93℃变性,65℃左右退火与延伸)。
4.出现片状拖带或涂抹带
PCR扩增有时出现涂抹带或片状带或地毯样带。其原因往往由于酶量过多或酶的质量差,dNTP浓度过高,Mg2+浓度过高,退火温度过低,循环次数过多引起。其对策有:①减少酶量,或调换另一来源的酶;②减少dNTP的浓度;③适当降低Mg2+浓度;④增加模板量,减少循环次数。
第三节本次竞赛PCR扩增反应的试剂盒说明
采用天根(TIANGEN,天根生化科技有限公司)2×HosStart Taq Master Mix,该产品包含HotStart Taq DNA 聚合酶、dNTPs、MgCl2、反应缓冲液、PCR反应增强剂和优化剂以及稳定剂,浓度为2×。
多重PCR原理
多重PCR原理: 一般PCR仅应用一对引物,通过PCR扩增产生一个核酸片段,主要用于单一致病因子等的鉴定.多重PCR(multiplex PCR),又称多重引物PCR或复合PCR,它是在同一PCR反应体系里加上二对以上引物,同时扩增出多个核酸片段的PCR反应,其反应原理,反应试剂和操作过程与一般PCR相同. 多重PCR的主要用于多种病原微生物的同时检测或鉴定和病原微生物,某些遗传病及癌基因的分型鉴定。多种病原微生物的同时检测或鉴定,是在同一PCR反应管中同时加上多种病原微生物的特异性引物,进行PCR扩增.可用于同时检测多种病原体或鉴定出是那一型病原体感染,,可系统组合的有:①肝炎病毒的感染,在同一病人或同一供血者体内,有时存在多种肝炎病毒重叠感染,有时是甲乙丙型肝炎病毒重叠;有时可能是甲乙型肝炎病毒重叠;有时是乙丙型肝炎病毒重叠.②肠道致病性细菌的检测,如伤寒,痢疾和霍乱,有时具有较相同的肠道症状,有时痢疾霍乱同存一病人并同时发病.③性病的检测,如梅毒,淋病及艾滋病的诊断.④战伤细菌及生物战剂细菌的检测,如破伤风杆菌,产气荚膜杆菌,炭疽杆菌,鼠疫杆菌等侦检.⑤需特殊培养的无芽胞厌氧菌,如脆弱类杆菌、艰难杆菌的鉴定等. 某些病原微生物,某些遗传病或癌基因,型别较多,或突变或缺失存在多个好发部位,多重PCR可提高其检出率并同时鉴定其型别及突变等可系统应用的有:乙型肝炎病毒的分型;乳头瘤病毒的分型;单纯疱疹病毒的分型;杜氏肌营养不良症的分型及癌基因的检测等. 多重PCR的特点有:①高效性,在同一PCR反应管内同时检出多种病原微生物,或对有多个型别的目的基因进行分型,特别是用一滴血就可检测多种病原体.②系统性,多重PCR很适宜于成组病原体的检测,如肝炎病毒,肠道致病性细菌,性病,无芽胞厌氧菌,战伤感染细菌及细菌战剂的同时侦检.③经济简便性,多种病原体在同一反应管内同时检出,将大大的节省时间,节省试剂,节约经费开支,为临床提供更多更准确的诊断信息. 试验准备: PCR 反应体积为 25μL ,包括:灭菌的超纯水、 PCR 缓冲液 (1x) 、 dNTP 混合物 (200μM each) 、引物 (0.04–0.6μM each); DMSO, 甘油或 BSA (5%); Taq DNA 聚合酶 (1–2 U/25μL) 和基因组DNA 模板 (150ng/25μL) 。先加入水,其它组分可以任意顺序加入,加样在冰上进行,加样完成后反应管直接从冰上转入预热到 94 ℃的循环仪加热模块或水浴中。对于放射性标记的反应,在反应开始前立即在100μL 的反应混合液中加入 1 μCi dNTP。使用 100μL 、 25μL 、
分子生物学综合实验报告
分子生物学综合试验报告
综合实验Ⅰ.Southern杂交 (质粒DNA提取、PCR技术体外扩增DNA、质粒载体和外源DNA的连接反应、 地高辛标记的Southern杂交) 一.实验目的 1.学习Southern杂交的原理及操作方法。 2.学习碱裂解法提取质粒的原理。 3.学习PCR反应的基本原理和实验技术;了解引物设计的一般要求。 4.掌握DNA体外连接的基本技能,了解连接反应的注意事项。 二.实验原理 利用染色体DNA与质粒DNA的变性与复性的差异而达到分离的目的。在碱变性条件下,染色体DNA的氢键断裂,双螺旋解开而变性,质粒DNA氢键也大部分断裂,双螺旋也有部分解开,但共价闭合环状结构的两条互补链不会完全分离,当pH=的乙酸钠将其pH调到中性时,变性的质粒DNA又恢复到原来的碱裂解法提取质粒的主要原理是:利用染色体DNA与质粒DNA的变性与复性的差异而构型,而染色体DNA不能复性,形成缠绕的致密网状结构,离心后,由于浮力密度不同,染色体DNA与大分子RNA、蛋白质-SDS复合物等一起沉淀下来而被除去。 聚合酶链反应(PCR)是体外酶促合成DNA片段的一种技术,PCR 进行的基本条件:DNA模板(在RT-PCR中模板是RNA)、引物、dNTP (dATP、dTTP、dGTP、dCTP)、Taq DNA聚合酶、反应缓冲体系。 PCR循环由三个步骤组成:变性、退火、延伸。每一个循环的产物可作为下一个循环的模板,通过30个左右循环后,目的片段的扩增可达106倍。
DNA片段之间的连接是通过DNA连接酶的催化实现的。DNA连接酶催化具有平末端或互补粘性末端的DNA片段间相邻碱基通过3’,5’磷酸二酯键连接起来。最常用的来源于T4噬菌体的T4DNA连接酶。对于平末端或互补的粘性末端可直接进行连接反应。一个片段是平末端,另一片段为粘性末端或两个片段都是粘性末端但不配对,则需要通过各种方式使其可一匹配或通过平末端进行连接。通常采用末端补平、加同聚物尾、加接头等方式是目的片段之间能够匹配。 地高辛随机引物法标记的原理:在随机引物法标记的反应液中,有随机合成的六聚核苷酸作为引物,dATP、dCTP、dGTP、dTTP和D1G-11-dUTP作为合成底物,以单链DNA作为模板,在Klenow酶的作用下,合成插入地高辛的DNA链。以地高辛标记的探针与靶基因DNA链杂交后,再通过免疫反应进行检测。一般通过酶标记地高辛抗体检测,就可以肯定杂交反应的存在。免疫检验一般用碱性磷酸酶系统,BClP/NBT显色,敏感性很高。 三.实验准备 1.实验材料: 含质粒的大肠杆菌DH5α,LB液体培养基, LB平板培养基 2.实验试剂: Taq DNA聚合酶,10×反应缓冲液(含25mmol MgCl2),dNTP,引物(P1、P2),溴乙啶 (EB) ,点样缓冲液Loading buffer(10×):%溴酚蓝,40%甘油,目的基因及载体, 2×ligation 缓冲液,T4 DNA连接酶, L CaCl2,氨苄青霉素(100mg/mL), TBE电泳缓冲液(5×), DIG Random Labeling Mix(高效),Anti-DIG-AP Conjugate, BCIP/NBT Stock Solution,Blocking Reagent。 20×SSC:柠檬酸钠,3M NaCl,2×SSC:柠檬酸钠, NaCl, EDTA,变性液: NaOH, NaCl,中和度: Tris-HCl、、3M NaCl,Standard buffer:5×SSC、%(w/v) N-Lauroylsarcosine, % (w/v) SDS, 1% Blocking Reagent,Standard buffer+50% formamide,Anti-DIG-AP 碱性磷酸酶标记抗地高辛单抗体,BCIP/NBT储备液,冲洗液:0. 1M
PCR扩增原理及操作
PCR扩增反应的操作 第一节PCR扩增反应的基本原理 一、聚合酶链式反应(PCR)的基本构成 PCR是聚合酶链式反应的简称,指在引物指导下由酶催化的对特定模板(克隆或基因组DNA)的扩增反应,是模拟体内DNA复制过程,在体外特异性扩增DNA片段的一种技术,在分子生物学中有广泛的应用,包括用于DNA作图、DNA测序、分子系统遗传学等。 PCR基本原理是以单链DNA为模板,4种dNTP为底物,在模板3’末端有引物存在的情况下,用酶进行互补链的延伸,多次反复的循环能使微量的模板DNA得到极大程度的扩增。在微量离心管中,加入与待扩增的DNA片段两端已知序列分别互补的两个引物、适量的缓冲液、微量的DNA 膜板、四种dNTP溶液、耐热Taq DNA聚合酶、Mg2+等。反应时先将上述溶液加热,使模板DNA 在高温下变性,双链解开为单链状态;然后降低溶液温度,使合成引物在低温下与其靶序列配对,形成部分双链,称为退火;再将温度升至合适温度,在Taq DNA聚合酶的催化下,以dNTP为原料,引物沿5’→3’方向延伸,形成新的DNA片段,该片段又可作为下一轮反应的模板,如此重复改变温度,由高温变性、低温复性和适温延伸组成一个周期,反复循环,使目的基因得以迅速扩增。因此PCR循环过程为三部分构成:模板变性、引物退火、热稳定DNA聚合酶在适当温度下催化DNA链延伸合成(见图)。 1.模板DNA的变性 模板DNA加热到90~95℃时,双螺旋结构的氢键断裂,双链解开成为单链,称为DNA的变性,以便它与引物结合,为下轮反应作准备。变性温度与DNA中G-C含量有关,G-C间由三个氢键连接,而A-T间只有两个氢键相连,所以G-C含量较高的模板,其解链温度相对要高些。故PCR 中DNA变性需要的温度和时间与模板DNA的二级结构的复杂性、G-C含量高低等均有关。对于高G-C含量的模板DNA在实验中需添加一定量二甲基亚砜(DMSO),并且在PCR循环中起始阶段热变性温度可以采用97℃,时间适当延长,即所谓的热启动。 2.模板DNA与引物的退火 将反应混合物温度降低至37~65℃时,寡核苷酸引物与单链模板杂交,形成DNA模板-引物复合物。退火所需要的温度和时间取决于引物与靶序列的同源性程度及寡核苷酸的碱基组成。一般要求引物的浓度大大高于模板DNA的浓度,并由于引物的长度显著短于模板的长度,因此在退火时,引物与模板中的互补序列的配对速度比模板之间重新配对成双链的速度要快得多,退火时间一般为1~2min。 3.引物的延伸 DNA模板-引物复合物在Taq DNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条与模板DNA链互补的新链。重复循环变性-退火-延伸三过程,就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。延伸所需要的时间取决于模板DNA的长度。在72℃条件下,Taq DNA聚合酶催化的合成速度大约为40~60个碱基/秒。经过一轮“变性-退火-延伸”循环,模板拷贝数增加了一倍。在以后的循环中,新合成的DNA都可以起模板作用,因此每一轮循环以后,DNA拷贝数就增加一倍。每完成一个循环需2~4min,一次PCR经过30~40次循环,约2~3h。扩增初期,扩增的量呈直线上升,但是当引物、模板、聚合酶达到一定比值时,酶的催化反应趋于饱和,便出现所谓的“平台效应”,即靶DNA产物的浓度不再增加。 PCR的三个反应步骤反复进行,使DNA扩增量呈指数上升。反应最终的DNA扩增量可用Y =(1+X)n计算。Y代表DNA片段扩增后的拷贝数,X表示平(Y)均每次的扩增效率,n代表循环次数。平均扩增效率的理论值为100%,但在实际反应中平均效率达不到理论值。反应初期,
生物实验4 实时定量PCR 实验报告
实验四定量PCR扩增 姓名:李宗翰专业:环境工程学号:1432999 同组人姓名:刘雪飞 一、实验目的 复习定量PCR原理,熟悉绝对定量的操作流程 二、实验原理 实时荧光定量PCR是在PCR反应体系中加入荧光基团,利用荧光信号累积实时监测整PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。在PCR扩增的指数时期,模板的Ct 值和该模板的起始拷贝数存在线性关系,通过Ct值和标准曲线的分析对起始模板进行定量分析 三、实验仪器及材料 real time PCR仪(ABI7500, RotorGene 3000),微量移液器,Tip头,0.2ml光学薄壁管,8联PCR管,1.5ml离心管,SYBR Mix,引物及108 copy/ul 标准品 四、实验步骤 1、标准样品稀释:取4个1.5ml的离心管,写上标记107,106, 105, 104,向每管加入90μl ddH2O,取10μl 108copy/ul 的标样加入到107管中,充分混匀后,从管中取10μl 107copy/ul的液体到106管中。按上述操作依次稀释,得到5个倍数的标准样品。注意,每次稀释都要换Tip头。 2、配制预混液:取119μl ddH2O、7μl引物4204f、7μl引物4448r和7μl Rox 到装有175μl SYBR Mix液的1.5ml离心管中,混匀。 3、分装预混液:取7个小离心管,分别标记108、107、106、105、10 4、UNK (未知样)、NTC(阴性对照)。向其中分别加入42μl预混液和4.7μl模板(1-5号加标样、6号加未知样、7号加等量ddH2O),混匀。 4、戴上手套取两个8联管,并排放置管架上。分别取上述样品20μl至第1-7管中(第8管空出),每个样品两个重复,共14个样品。将加好样的8联管振荡。 5、设置分析仪参数如下:95℃3min;95℃15s+60℃40s为一个循环,循环次数40,融解曲线温度范围60~95℃。振荡好的样品进机进行PCR扩增。 6、扩增后利用软件分析C T值及未知样的定量结果。
DNA提取及PCR扩增实验报告.doc
PCR扩增及DNA琼脂糖凝胶电泳 刘琳1131428 环境科学 一、实验目的 1.学习并掌握PCR扩增的基本原理与实验技术。 2.对扩增后的DNA进行琼脂糖凝胶电泳试验,并分析相应结果。 二、实验原理 1. PCR扩增 多聚酶链反应(PCR)技术的原理类似于DNA的天然复制过程。在微量离心管中加入适量缓冲液,加入微量模板DNA、四种脱氧核苷酸(dNTP)、耐热T aq聚合酶及两个合成DNA的引物,而后加热使模板DNA在高温下(94℃)变性,双链解链,这是所谓变性阶段。降低溶液温度,使合成引物在低温(55℃)与模板DNA互补退火形成部分双链,这是所谓退火阶段。溶液反应温度升至中温(72℃),在Tap酶作用下,用四种dNTP为原料,引物为复制起点,模板DNA的一条双链在解链和退火之后延伸为两条双链,这是延伸阶段。如此反复,在同一反应体系中可重复高温变性、低温退火和DNA合成这一循环,使产物DNA重复合成,并在重复过程中,前一循环的产物DNA可作为后一循环的模板DNA而参与DNA的合成,使产物DNA的量按指数方式扩增。经过30~40个循环,DNA扩增即可完成。 2. DNA琼脂糖凝胶电泳实验 DNA分子在高于其等电点的溶液中带负电,在电场中向阳极移动。在一定的电场强度下,DNA分子的迁移速度取决于分子筛效应,即分子本身的大小和构型是主要的影响因素。DNA分子的迁移速度与其相对分子量成反比。不同构型的DNA分子的迁移速度不同。该电泳方法以琼脂凝胶作为支持物,利用DNA分子在泳动时的电荷效应和分子筛效应,达到分离混合物的目的。 三、实验材料 仪器:PCR扩增仪、0.2ul薄壁管、1.5ml离心管、移液枪、枪头、微波炉、电泳仪、水平电泳槽、制胶版、紫外透射仪。 试剂:TapDNA聚合酶、dNTP、buffer、两种引物、16S全长DNA样本、无菌ddH2O、模板DNA 、TBE、琼脂糖、EB、显色剂。 四、实验步骤 1. PCR扩增 本次试验选择细菌16S rDNA V3区片段进行扩增。 1.1 根据计算,首先取1.5ml离心管按照 2.5ul 10×Buffer 、1 ul dNTP、0.5 ul 341GC、 0.5 ul 534、0.125 ul Taq、19.375u ddH2O的比例配置足量的PCR反应体系。 1.2 分别向9个薄壁管中分别加入24 ul的反应体系,并分别添加8种不同的模版,并于第9个薄壁管中加入无菌ddH2O作为阴性对照。 1.3 将薄壁管放入PCR扩增仪中,按照预定程序进行PCR扩增。其中循环过程需要达到30~40次。程序如下: 预变性:94℃3min 循环:94℃变性30s 55℃退火30s 72℃延伸30s 末次延伸:72℃5min
pcr扩增的原理和步骤
PCR 扩增的原理和步骤 聚合酶链反应(polymerasechain reaction PCR)技术是20世纪80年代中期发 展起来的一项基因检测即一种体外核酸扩增技术。它具有许多优点:特异性、易重复、高效性等,可以在几个小时完成过去几天或者更长时间完成的实验,因此这项技术在生物医学领域具有划时代的意义。但是,传统PCR技术有它的缺点,它通过电泳对扩增反应的最终产物进行定性分析而不能对起始模板准确定量,同时也无法对扩增反应实时检测且在实验过程中易污染而出现假阳性。人们为了寻找更为灵敏、快速、简便、高特异性的方法进行了许多探索研究,直到1996 年由美国Applied Biosystems公司推出了一种新的定量试验技术—荧光定量PCR(F lurogenic Quantitative Polymerase Chain Reaction,FQ-PCR;real-time quantitati ve PCR,RT-qPCR or qPCR),它是通过荧光染料或荧光标记的特异性探针,标记 跟踪PCR产物进行实时监测反应,利用与之相适应的软件对产物进行分析,计 算待测样品模板的初始浓度,实现了PCR 从定性到定量质的跨越,具有里程碑 意义。目前,此项技术已应用于干细胞研究、肿瘤学和遗传疾病研究、病原体检 测和传染病研究、药物分析、药物基因组学、植物学研究和农业生物科技等多领 域研究中。本文对实时荧光定量PCR 的原理、分类和应用进行阐述。 一、实时荧光定量PCR技术的原理 real-time quantitative PCR 技术是指在 PCR 反应体系中加入荧光基团,通过 荧光信号不断累积而实现实时监测PCR全程,然后通过标准曲线对未知模板进 行定量分析的方法。在荧光定量PCR技术中有2个概念比较重要。(1)荧光域值(t hreshold)的设定: PCR 反应的前 15 个循环的荧光信号作为荧光本底信号,荧光域值的缺省设置是3~15 个循环的荧光信号标准偏差的10 倍。(2)Ct 值:C 代表Cycle,t 代表 threshold,Ct 值的含义是每个反应管内的荧光信号到达设定的域值 时所经历的循环数。在实时荧光定量PCR中,对全程PCR扩增过程进行实时检测,根据反应时间和荧光信号的变化可以绘制成一条曲线。一般来说,整条曲线可以分3个阶段:荧光背景信号阶段、荧光信号指数扩增阶段和平台期。在荧光背景信号阶段,扩增的荧光信号与背景无法区分,无法判断产物量的变化。在平台期,扩增产物已不再呈指数级的增加,所以反应终产物量与起始模板量之间已 经不存在线性关系,通过反应终产物也算不出起始DNA 拷贝数。只有在荧光产
RT-PCR实验报告课件
逆转录pcr rt-pcr 为反转录rcr (reverse transcription pcr )和实时pcr (real time pcr )共同的缩写。逆转录pcr ,或者称反转录pcr(reverse transcription-pcr, rt-pcr) ,是聚合酶链式反应(pcr) 的一种广泛应用的变形。在rt-pcr 中,一条rna 链被逆转录成为互补dna,再以此为模板通过pcr 进行dna 扩增。 由一条rna 单链转录为互补dna(cdna) 称作“逆转录”,由依赖rna 的dna 聚合酶(逆转录酶)来完成。随后,dna 的另一条链通过脱氧核苷酸引物和依赖rna 的dna 聚合酶完成,随每个循环倍增,即通常的pcr 。原先的rna 模板被rna 酶h 降解,留下互补dna。 rt-pcr 的指数扩增是一种很灵敏的技术,可以检测很低拷贝数的rna 。rt-pcr 广泛应用于遗传病的诊断,并且可以用于定量监测某种rna 的含量。(检测基因表达的方法,参见northern blot 法。) rt-pcr 有时候也会指代实时pcr(real-time pcr) 。为了与逆转录pcr 相区别,通常被写作“定量pcr ”(quantitative pcr) 或者rtq-pcr(real-time quantitative pcr) 。 实时pcr 实时pcr(real-time pcr) ,属于定量pcr (q-pcr )的一种,以一定时间内dna 的增幅量为基础进行dna 的定量分析。real time pcr 的定量使用萤光色素,目前有二种方法。 一种是在ds dna 中插入特异的萤光色素;另一种使用一种能与增幅dna 序列中特定寡核酸序 列相结合的一种萤光探针(probe )。real time pcr 与reverse transcription pcr 相结合,能用微量的rna 来找出特定时间、细胞、组织内的特别表达的遗传基因。这两种rt pcr 的组合又被称之为“定量rt-pcr (quantitative rt-pcr )” rt-pcr 技术相关试剂 oligo: 多聚体,相当于mrna 引物 amv (m-mlv):逆转录酶 dntp :脱氧核苷酸 rnase :rna 酶抑制剂 pcr buffer :rt-pcr 缓冲液 mgcl2 :2 价镁离子 pcr 各步骤的目的 (一)预变性: 破坏dna 中可能存在的较难破坏的二级结构。使dna 充分变性,减少dna 复杂结构对扩增的影响,以利于引物更好的和模板结合,特别是对于基因组来源的dna 模板,最好不要吝 啬这个步骤。此外,在一些使用热启动taq 酶的反应中,还可激活taq 酶,从而使pcr 反应得以顺利进行。 (二)变性-- 退火-- 延伸循环: ①模板dna 的变性:模板dna 经加热至93℃左右一定时间后,使模板dna 双链或经pcr 扩增形成的双链dna 解离,使之成为单链,以便它与引物结合,为下轮反应作准备; ②模板dna 与引物的退火( 复性) :模板dna 经加热变性成单链后,温度降至55℃左右,引物与模板dna 单链的互补序列配对结合; ③引物的延伸:dna 模板-- 引物结合物在taqdna 聚合酶的作用下,以dntp 为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板dna 链互补的半保留复制链。 (三)pcr 仪扩增循环后72 度延伸10 分钟 用pcr 仪扩增时,( 变性. 退火, 延伸) 循环完成后, 继续72 度延伸了10 分钟的原因:
PCR实验报告
PCR实验报告 7月19日高遄 实验目的:了解PCR技术原理,掌握最基础的PCR实验步骤。 实验试剂:模板DNA,Mg2+,buffer,dNTPs,Taq DNA聚合酶,引物,H2O,石蜡油。 实验原理: ●PCR全称聚合酶链反应,是体外快速扩增特定基因或DNA序列最常用的方法。 ●基本原理:首先将双链DNA分子在临近沸点的温度下加热分离成2条单链DNA分子,DNA聚合酶以单链DNA为模板并利用反应混合物中的四种脱氧核苷三磷酸合成新的DNA互补链。PCR反应时,只要在试管内加入模板DNA、PCR引物、四种核苷酸及适当浓度的Mg2+,DNA聚合酶就能在数小时内将目标序列扩增100万倍以上。 (1)双链模板DNA分子首先在高温下解开成长的单链,短链引物分子立即与该模板DNA 两端的特定序列相结合,产生双链区。 (2)DNA聚合酶从引物处开始复制其互补链,迅速产生与目标序列完全相同的复制品。(3)在后续反应中,无论是起始模板DNA还是经复制的杂合DNA双链,都会在高温下解开成为单链,体系中的引物分子再次与其互补序列相结合,聚合酶也再度复制模板 DNA。 (4)由于在PCR反应中选用的一对引物,是按照与扩增区域两端序列彼此互补的原则设计的,因此每一条新生链的合成都是从引物的退火结合位点开始并朝反方向延伸的,每一条新合成的DNA链上都有新的引物结合位点。 (5)整个PCR的反应全过程,即DNA解链(变性)、引物与模板DNA结合(退火)、DNA 合成(链的延伸)三步可以被不断重复。经多次循环之后,反应混合物中所含有的双链DNA分子数,即两条引物结合位点之间的DNA区段的拷贝数,理论上的最高值应该是2^n,能进一步满足遗传分析的需要。 ●试剂作用: (1)引物:DNA复制的起始点,针对复制DNA片段的两端,有5’引物和3’引物 (2)Taq DNA聚合酶:促进dNTPs与模板结合。 (3)Buffer:Tris-HCl反应缓冲液,Taq DNA聚合酶提供一个最适酶催反应条件。 (4)Mg2+:对PCR扩增效率影响很大,浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。 (5)dNTPs:底物,在引物引导下合成与模板互补的DNA新链。 (6)石蜡油:防止PCR加热过程中DNA蒸发。 ●试剂配置体积: (1)热启动:94℃,5~10min (2)变性:94℃,45~60s。 (3)退火:50~65℃,1min。退火温度计算:Tm-(5℃~10℃),Tm(解链温度)=4(G+C)+2(A+T)。 (4)延伸:72℃,1~1.5min。 (5)步骤(2)~(4)热循环25~30个周期 (6)保温:延伸72℃,10min ●产物检测:凝胶电泳。
PCR扩增的原理和步骤
PCR扩增的原理和步骤 聚合酶链反应(polymerasechain reaction PCR)技术是20世纪80年代中期发展起来的一项基因检测即一种体外核酸扩增技术。它具有许多优点:特异性、易重复、高效性等,可以在几个小时完成过去几天或者更长时间完成的实验,因此这项技术在生物医学领域具有划时代的意义。但是,传统PCR技术有它的缺点,它通过电泳对扩增反应的最终产物进行定性分析而不能对起始模板准确定量,同时也无法对扩增反应实时检测且在实验过程中易污染而出现假阳性。人们为了寻找更为灵敏、快速、简便、高特异性的方法进行了许多探索研究,直到1996年由美国Applied Biosystems公司推出了一种新的定量试验技术—荧光定量PCR(F lurogenic Quantitative Polymerase Chain Reaction,FQ-PCR;real-time quantitati ve PCR,RT-qPCR or qPCR),它是通过荧光染料或荧光标记的特异性探针,标记跟踪PCR产物进行实时监测反应,利用与之相适应的软件对产物进行分析,计算待测样品模板的初始浓度,实现了PCR从定性到定量质的跨越,具有里程碑意义。目前,此项技术已应用于干细胞研究、肿瘤学和遗传疾病研究、病原体检测和传染病研究、药物分析、药物基因组学、植物学研究和农业生物科技等多领域研究中。本文对实时荧光定量PCR的原理、分类和应用进行阐述。 一、实时荧光定量PCR技术的原理 real-time quantitative PCR技术是指在PCR反应体系中加入荧光基团,通过荧光信号不断累积而实现实时监测PCR全程,然后通过标准曲线对未知模板进行定量分析的方法。在荧光定量PCR技术中有2个概念比较重要。(1)荧光域值(t hreshold)的设定:PCR反应的前15个循环的荧光信号作为荧光本底信号,荧光域值的缺省设置是3~15个循环的荧光信号标准偏差的10倍。(2)Ct值:C代表Cycle,t代表threshold,Ct值的含义是每个反应管内的荧光信号到达设定的域值时所经历的循环数。在实时荧光定量PCR中,对全程PCR扩增过程进行实时检测,根据反应时间和荧光信号的变化可以绘制成一条曲线。一般来说,整条曲线可以分3个阶段:荧光背景信号阶段、荧光信号指数扩增阶段和平台期。在荧光背景信号阶段,扩增的荧光信号与背景无法区分,无法判断产物量的变化。在平台期,扩增产物已不再呈指数级的增加,所以反应终产物量与起始模板量之间已经不存在线性关系,通过反应终产物也算不出起始DNA拷贝数。只有在荧光产生进入指数期,PCR产物量的对数值与起始模板量之间存在线性关系,所以在P CR反应处于指数期的某一点上来检测PCR产物的量,由此来推断模板最初的含
PCR各步骤的目的
P C R各步骤的目的 Prepared on 22 November 2020
PCR各步骤的目的 (一)预变性: 破坏DNA中可能存在的较难破坏的二级结构。使DNA充分变性,减少DNA复杂结构对扩增的影响,以利于引物更好的和模板结合,特别是对于基因组来源的DNA模板,最好不要吝啬这个步骤。此外,在一些使用热启动Taq酶的反应中,还可激活Taq酶,从而使PCR反应得以顺利进行。 (二)变性--退火--延伸循环: ①模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备; ②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合; ③引物的延伸:DNA模板--引物结合物在TaqDNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链。 (三)用PCR仪扩增时,(变性.退火,延伸)循环完成后,继续72度延伸了10分钟的原因: 1.延伸时间取决于待扩增DNA片段的长度。(当然是在反应体系一定的条件下)例如,使用taq DNA聚合酶,72度时的碱基掺入率为35-100bp/s,因此延伸速率为1kb/min。 2.根据延伸速率推得,扩增1kb以内的dna片段1min即可,而3-4kb则需要3-4min,依次照推。通常在最后一轮要适当的将延伸时间延长至4-10min,这样做是使pcr反应完全以提高扩增产量。 3.继续72度延伸了10分钟除了可以使pcr反应完全以提高扩增产量外,还有一个作用是:在用普通taq酶进行PCR扩增时在产物末端加A尾的作用,可以直接用于TA克隆的进行。
实验二PCR扩增(聚合酶链式反应)
实验二 PCR扩增(聚合酶链式反应) 一、实验目的 1.学习聚合酶链式反应概念及技术方法; 2.掌握聚合酶链式反应操作过程。 二、实验原理(聚合酶链式反应) PCR是聚合酶链式反应,是以单链DNA为模板,4种dNTP为底物,在模板3’末端有引物存在的情况下,用酶进行互补链的延伸,多次反复的循环能使微量的模板DNA得到极大程度的扩增。在微量离心管中,加入与待扩增的DNA片段两端已知序列分别互补的两个引物、适量的缓冲液、微量的DNA膜板、四种dNTP溶液、耐热Taq DNA聚合酶、Mg2+等。 反应时先将上述溶液加热,使模板DNA在高温下变性,双链解开为单链状态;然后降低溶液温度,使合成引物在低温下与其靶序列配对,形成部分双链,称为退火;再将温度升至合适温度,在Taq DNA聚合酶的催化下,以dNTP为原料,引物沿5’→3’方向延伸,形成新的DNA片段,该片段又可作为下一轮反应的模板,如此重复改变温度,由高温变性、低温复性和适温延伸组成一个周期,反复循环,使目的基因得以迅速扩增。因此PCR循环过程为三部分构成:模板变性、引物退火、热稳定DNA聚合酶在适当温度下催化DNA链延伸合成 1.模板DNA的变性 模板DNA加热到90-95℃时,双螺旋结构的氢键断裂,双链解开成为单链,称为DNA的变性,以便它与引物结合,为下轮反应作准备。变性温度与DNA中G-C含量有关,G-C间由三个氢键连接,而A-T间只有两个氢键相连,所以G-C含量较高的模板,其解链温度相对要高些。故PCR中DNA变性需要的温度和时间与模板DNA的二级结构的复杂性、G-C含量高低等均有关。对于高G-C含量的模板DNA在实验中需添加一定量二甲基亚砜(DMSO),并且在PCR循环中起始阶段热变性温度可以采用97℃,时间适当延长,即所谓的热启动。 2.模板DNA与引物的退火 将反应混合物温度降低至37-65℃时,寡核苷酸引物与单链模板杂交,形成DNA模板-引物复合物。退火所需要的温度和时间取决于引物与靶序列的同源性程度及寡核苷酸的碱基组成。一般要求引物的浓度大大高于模板DNA的浓度,并由于引物的长度显著短于模板的长度,因此在退火时,引物与模板中的互补序列的配对速度比模板之间重新配对成双链的速度要快得多,退火时间一般为1-2min。 3.引物的延伸 DNA模板-引物复合物在Taq DNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条与模板DNA链互补的新链。重复循环变性-退火-延伸三过程,就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。延伸所需要的时间取决于模板DNA的长度。在72℃条件下,Taq DNA聚合酶催化的合成速度大约为40-60个碱基/秒。经过一轮“变性-退火-延伸”循环,模板拷贝数增加了一倍。在以后的循环中,新合成的DNA都可以起模板作用,因此每一轮循环以后,DNA拷贝数就增加一倍。每完成一个循环需2-4min,一次PCR经过30-40次循环,约2-3h。扩增初期,扩增的量呈直线上升,但是当引物、模板、聚合酶达到一定比值时,酶的催化反应趋于饱和,便出现所谓的“平台效应”,即靶DNA产物的浓度不再增加。
PCR扩增实验操作步骤
PCR扩增反应 一、实验原理 PCR:是一种选择性扩增DNA或RNA的方法,其基本原理是依据体内细胞分裂中的DNA半保留复制机理,以及在体外dNTP分子于不同温度下双链和单链可以互相转变的性质,人为地控制体外合成系统的温度,以促使双链DNA变成单链DNA;单链DNA与人工合成的引物退火,以及在dNTP存在下,耐高温的DNA聚合酶使引物沿单链模板延伸成为双链DNA。 PCR反应分3步:①变性:通过加热使DNA 双螺旋的氢键断裂,双链解离形成单链DNA;②退火:当温度突然降低时,由于模板分子结构较引物要复杂得多,而且反应体系中引物DNA量大大多于模板DNA,使引物和其互补的模板在局部形成杂交链,而模板DNA 双链之间互补的机会较少。③延伸:在DNA聚合酶和4 种dNTP底物及Mg2+存在的条件下,5'→3'的聚合酶催化以引物为起始点的DNA链延伸反应,以上3步为一个循环,每一循环的产物可以作为下一个循环的模板,数小时之后,介于两个引物之间的特异性DNA片段得到了大量复制,数量可达2×106~7拷贝。 变性 退火 延伸 图反应历程
二、实验材料 1·模板:细菌DNA 2·TsgDNA聚合酶3·dNTP混合液 4·10倍浓度PCR缓冲液5·2.5mmol/LMgCl2 6·RAPD引物:S14 S15 S18 S66 S74 S88 S97 S103 S110 S115 7·提取细菌DNA的相关试剂 三、操作步骤 1.细菌染色体DNA的提取(见上一组) 3·反应程序: 将RAPD反应试剂加入EP管中 轻混后用100ul石蜡油覆盖于反应混合液之上,防止样品在反复加热-冷却的过程中蒸发,盖好盖子 打开PCR反应仪输入以下反应数据 ●94 摄氏度预变性5min ●94摄氏度变性40s ●40摄氏度退火40s ●72摄氏度延伸1min 将EP管放入仪器开始扩增,循环35次;72摄氏度延伸10min 仪器为Model MyGene 25 Plus 三、注意事项 1、PCR反应体系中DNA样品及各种试剂的用量都极少,必须严格注意吸样量的准确性 及全部放入反应体系中。 2、为避免污染凡是用在PCR反应中的Tip尖、离心管、蒸馏水都要灭菌;吸每种试剂 时都要换新的灭菌Tip尖。 3、加试剂时先加消毒三蒸水,最后加DNA模板和Taq DNA聚合酶。 4、置PCR仪进行PCR反应前,PCR管要盖紧,否则使液体蒸发影响PCR反应。 5.引物条件首先引物与模板的序列要紧密互补,其次引物与引物之间避免形成稳定二聚 体或发夹结构,再次引物不能在模板的非目的位点引发DNA聚合反应(即错配)。
PCR扩增反应的操作实验报告1
PCR扩增反应的操作实验报告 实验原理: 以单链DNA为模板,4种dNTP为底物,在模板3’末端有引物存在的情况下,用酶进行互补链的延伸,多次反复的循环能使微量的模板DNA得到极大程度的扩增。在微量离心管中,加入与待扩增的DNA片段两端已知序列分别互补的两个引物、适量的缓冲液、微量的DNA膜板、四种dNTP溶液、耐热Taq DNA聚合酶、Mg2+等。反应时先将上述溶液加热,使模板DNA在高温下变性,双链解开为单链状态;然后降低溶液温度,使合成引物在低温下与其靶序列配对,形成部分双链,称为退火;再将温度升至合适温度,在Taq DNA 聚合酶的催化下,以dNTP为原料,引物沿5’→3’方向延伸,形成新的DNA片段,该片段又可作为下一轮反应的模板,如此重复改变温度,由高温变性、低温复性和适温延伸组成一个周期,反复循环,使目的基因得以迅速扩增。因此PCR循环过程为三部分构成:模板变性、引物退火、热稳定DNA聚合酶在适当温度下催化DNA链延伸合成。 实验步骤:一、PCR反应的条件 PCR反应条件为温度、时间和循环次数。 1.温度与时间的设置 (1)变性温度与时间:变性温度低,解链不完全是导致PCR失败的最主要原因。 一般情况下,93℃~94℃ lmin足以使模板DNA变性,若低于93℃则需延长时间,但温度不能过高,因为高温环境对酶的活性有影响。此步若不能使靶基因模板或PCR产物完全变性,就会导致PCR失败。 (2)退火(复性)温度与时间:退火温度是影响PCR特异性的较重要因素。变性后温度快速冷却至40℃~60℃,可使引物和模板发生结合。由于模板 DNA比引物复杂得多,引物和模板之间的碰撞结合机会远远高于模板互补链之间的碰撞。退火温度与时间,取决于引物的长度、碱基组成及其浓度,还有靶基序列的长度。对于20个核苷酸,G+C含量约50%的引物,55℃为选择最适退火温度的起点较为理想。 (3)延伸温度与时间:Taq DNA聚合酶的生物学活性:70~80℃,150核苷酸/S/酶分子;70℃,60核苷酸/S/酶分子;55℃,24核苷酸/S/酶分子;高于90℃时,DNA合成几乎不能进行。 (4)PCR反应的延伸温度一般选择在70~75℃之间,常用温度为72℃,过高的延伸温度不利于引物和模板的结合。PCR延伸反应的时间,可根据待扩增片段的长度而定,一般1kb以内的DNA片段,延伸时间1min是足够的。 3~4kb的靶序列需3~4min;扩增10kb需延伸至15min。延伸进间过长 会导致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。 2.循环次数 循环次数决定PCR扩增程度。PCR循环次数主要取决于模板DNA的浓度,一般的循环次数选在30~40次之间,循环次数越多,非特异性产物的量亦随之增多。 二、PCR扩增产物分析 PCR产物是否为特异性扩增,其结果是否准确可靠,必须对其进行严格的分析与鉴定,才能得出正确的结论。PCR产物的分析,可依据研究对象和目的不同而采用不同的分析方法。
pcr扩增的原理和步骤
pcr扩增的原理和步骤 一、基本原理:PCR技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。DNA的半保留复制是生物进化和传代的重要途径。双链DNA在多种酶的作用下可以变性解旋成单链,在DNA聚合酶的参与下,根据碱基互补配对原则复制成同样的两分子拷贝。 在实验中发现,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,加入设计引物,DNA 聚合酶、dNTP就可以完成特定基因的体外复制。 二、PCR由变性--退火--延伸三个基本反应步骤构成: 1、模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA 双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备。 2、模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合。 3、引物的延伸:DNA模板--引物结合物在72℃、DNA聚合酶(如TaqDNA 聚合酶)的作用下,以dNTP为反应原料,靶序列为模板,按碱基互补配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链。
重复循环变性--退火--延伸三过程就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。 扩展: 在实践中,聚合酶链式反应(PCR)可以因各种原因而失败,部分原因是由于其对于污染的敏感性,导致扩增错误的DNA产物。正因为如此,人们已经开发了一些技术和步骤来优化聚合酶链式反应条件。将聚合酶链式反应前的混合物与潜在DNA污染物分开的实验室方案和流程解决了外源DNA的污染问题。 这通常包括从用于分析的区域分理出聚合酶链式反应的设定区域或者说聚合酶链式反应产物的纯化,一次性塑料制品的使用,及对反应装置之间的工作台面彻底清洁。引物的设计技术在改善聚合酶链式反应产物产率和避免杂产物的形成是很重要的。
PCR实验报告
PCR实验报告 7月19日高遗 实验目的:了解PCF技术原理,掌握最基础的PCF实验步骤。 实验试剂:模板DNA Mg2+ buffer,dNTPs Taq DNA聚合酶,引物,H2Q石蜡油。实验原理:PCF全称聚合酶链反应,是体外快速扩增特定基因或DNAJ列最常用的方法。 基本原理:首先将双链DNA分子在临近沸点的温度下加热分离成2条单链DNA分子,DNA聚合酶以单链DNA为模板并利用反应混合物中的四种脱氧核苷三磷酸合成新的DNA互补链。PCF反应时,只要在试管内加入模板DNAPCF引物、四种核苷酸及适当浓度的Mg2+ DNA聚合酶就能在数小时内将目标序列扩增100万倍以上。 (1)双链模板DNA分子首先在高温下解开成长的单链,短链引物分子立即与该模板DNA 两端 的特定序列相结合,产生双链区。 (2)DNA聚合酶从引物处开始复制其互补链,迅速产生与目标序列完全相同的复制品。 (3)在后续反应中,无论是起始模板DNA还是经复制的杂合DNA双链,都会在高温下解开 成为单链,体系中的引物分子再次与其互补序列相结合,聚合酶也再度复制模板DNA (4)由于在PCR反应中选用的一对引物,是按照与扩增区域两端序列彼此互补的原则设计 的,因此每一条新生链的合成都是从引物的退火结合位点开始并朝反方向延伸的,每一条新合成的DNA链上都有新的引物结合位点。 (5)整个PCR勺反应全过程,即DNA解链(变性)、弓I物与模板DNA吉合(退火)、DNA 合 成(链的延伸)三步可以被不断重复。经多次循环之后,反应混合物中所含有的双 链DNA分子数,即两条引物结合位点之间的DNA区段的拷贝数,理论上的最高值应该是25,能进一步满足遗传分析的需要。 试剂作用: (1)引物:DNA M制的起始点,针对复制DNA片段的两端,有5'引物和3'引物 (2)Taq DNA聚合酶:促进dNTPs与模板结合。 (3)Buffer : Tris-HCl反应缓冲液,Taq DNA聚合酶提供一个最适酶催反应条件。 (4)Mg2+:对PCF扩增效率影响很大,浓度过高可降低PCF T增的特异性,浓度过低则影 响PCF扩增产量甚至使PCF扩增失败而不出扩增条带。 (5)dNTPs:底物,在引物引导下合成与模板互补的DNA新链。 (6)石蜡油:防止PCF加热过程中DNA蒸发。 试剂配置体积: 温度和时间: (1) 热启动:94C,5~10min (2) 变性:94C,45~60s。 (3) 退火:50~65C,1min。退火温度计算:Tm- (5C~10C),Tm(解链温度)=4 (G+C +2 (A+T)o (4) 延伸:72C,1~1.5min。 (5) 步骤(2) ~ (4)热循环25~30个周期 (6) 保温:延伸72C,10min 产物检测:凝胶电泳。 实验步骤: (1)设计20卩l体系各试剂配比:
PCR的基本步骤及注意
(DNApolymerase I)最早于1955年发现,而较具有实验价值及实用性的Klenow fragment of E. Coli则是于70年代的初期由Dr. H. Klenow 所发现,但由于此酶不耐高温,高温能使之变性,因此不符合使用高温变性的聚合酶链式反应。现今所使用的酶(简称Taq polymerase), 则是于1976年从温泉中的(Thermusaquaticus)分离出来的。它的特性就在于能耐高温,是一个很理想的酶,但它被广泛运用则于80年代之后。PCR最初的原始雏形概念是类似修复复制,它是于1971年由 Dr. Kjell Kleppe提出。他发表了第一个单纯且短暂复制(类似PCR 前两个周期反应)的实验。而现今所发展出来的PCR则于1983由 Dr. Kary B. Mullis发展出的,Dr. Mullis当年服务于PE公司,因此PE公司在PCR界有着特殊的地位。Dr. Mullis 并于1985年与Saiki等人正式发表了第一篇相关的论文。此后,PCR的运用一日千里,相关的论文发表质量可以说是令众多其它研究方法难望其项背。随后PCR技术在生物科研和临床应用中得以广泛应用,成为分子生物学研究的最重要技术。Mullis也因此获得了1993年化学奖。 PCR原理 DNA的是生物进化和传代的重要途径。双链DNA在多种酶的作用下可以变性成单链,在DNA聚合酶的参与下,根据复制成同样的两分子挎贝。在实验中发现,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,加入设计,DNA 聚合酶、dNTP就可以完成特定基因的体外复制。 但是,DNA聚合酶在高温时会失活,因此,每次循环都得加入新的DNA聚合酶,不仅操作烦琐,而且价格昂贵,制约了PCR技术的应用和发展。 发现耐热DNA聚合酶--Taq酶对于PCR的应用有里程碑的意义,该酶可以耐受90℃以上的高温而不失活,不需要每个循环加酶,使PCR技术变得非常简捷、同时也大大降低了成本,PCR技术得以大量应用,并逐步应用于临床。 PCR技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性--退火--延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板--引物结合物在的作用下,以dNTP为反应原料,靶序列为模板,按与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链,重复循环变性--退火--延伸三过程就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟,2~3小时就能将待扩扩增放大几百万倍。 [PCR反应体系与反应条件] 1.标准的PCR反应体系 10×扩增缓冲液10μl 4种dNTP混合物200μl 引物10~100μl 模板DNA ~2μg